Préparation du milieu d’auto-induction : Le milieu doit être préparé avec les solutions stériles et l’équipement stérile obtenus à la semaine 1.
CONSEIL : Toutes ces étapes doivent être effectuées en utilisant une technique stérile avec une flamme allumée. Les pipettes et la paillasse doivent être nettoyées avec de l’eau et de l’éthanol avant de commencer. Assurez-vous d’utiliser uniquement des embouts, des fioles, des pipettes, de l’eau et des milieux stériles.
Préparation du milieu d’auto-induction pour l’expression (100 ml). Mettez à l’échelle pour obtenir la quantité dont vous avez besoin pour votre expérience en fonction des quantités suivantes pour 100 ml. Préparez un mélange frais et ne prévoyez pas de stocker le milieu auto-inductif (MAI) en excès.
Aspartate (5 %, pH 7,5) 5 ml
Glycérol (10 %) 5 ml
Sels minéraux 25x 4 ml
Glucose (40 %) 0,125 ml
MgSO4 (1 M) 0,2 ml
Arabinose (20 %) 0,25 ml
Métaux traces (5 000x) 20 µl
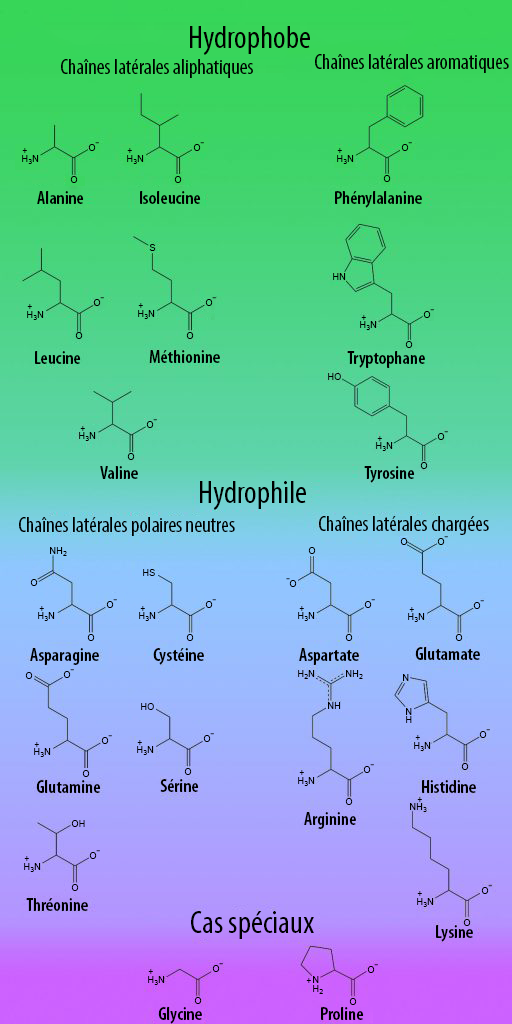
Mélange de 18 AA (25x) conservé à 4 °C 4 ml
Ajoutez les antibiotiques appropriés (ampicilline à une concentration finale de 100 µg/ml et tétracycline à 25 µg/ml).
Ajoutez de l’eau stérile pour obtenir un volume final de 100 ml.
Volumes et fioles recommandés pour les expressions pendant les semaines 2 et 3:
Protéine de type sauvage 75 ml Expression en fiole de 250 ml
Protéine ncAA 1 75 ml Expression en fiole de 250 ml
Protéine ncAA 1 (contrôle négatif -ncAA) 5 ml Expression en tube à culture
Protéine ncAA 2 75 ml Expression en fiole de 250 ml
Protéine ncAA 2 (contrôle négatif -ncAA) 5 ml Expression en tube à culture
Contrôle sfGFP sauvage (-ncAA) 5 ml Expression en tube à culture
CONSEIL (EXEMPLE POUR 250 ml — METTEZ À L’ÉCHELLE SELON VOS BESOINS): Il est pratique de commencer avec une bouteille de 250 ml d’eau stérile et d’en retirer environ 50 ml en les plaçant dans un autre récipient stérile (pour une utilisation ultérieure). Ensuite, tous les composants du milieu peuvent être ajoutés à 200 ml d’eau stérile et complétés avec de l’eau stérile à un volume final de 250 ml. Ensuite, ajoutez 250 microlitres d’ampicilline à 100 mg/ml au milieu, puis placez 75 ml de celui-ci dans une fiole avec déflecteur de 250 ml pour l’expression des protéines de type sauvage. 175 microlitres de tétracycline à 25 mg/ml peuvent ensuite être ajoutés au milieu restant (175 ml), qui peut ensuite être réparti dans les fioles d’expression. Les ncAA seront ajoutés plus tard.
Chaque section de laboratoire doit utiliser le code couleur de ruban approprié pour étiqueter les fioles afin qu’il soit facile de les récupérer. Rappelez-vous qu’il y a 4 sections et plus de 80 étudiants et, certaines semaines, 80 à 100 fioles en activité.
Ajout de cellules provenant de cultures de départ :
Dans les fioles préparées (75 ml de milieu par fiole), ajoutez des cultures cellulaires saturées en milieu non inductif de la lignée cellulaire appropriée pour commencer les expressions. Diluez les cellules appropriées issues des cultures de départ de la nuit du 1/100e au 1/200e dans chaque fiole contenant 75 ml. Incubez ces cultures pendant 0,5 à 1 heure avant d’ajouter les ncAA (cette incubation peut être raccourcie pour certains ncAA).
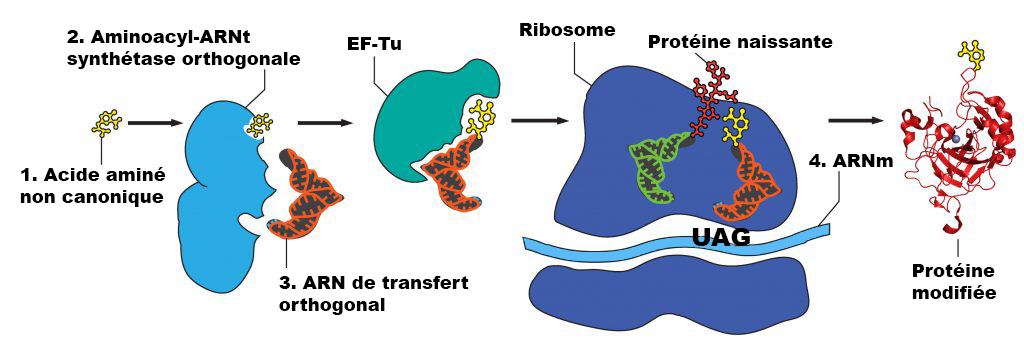
Préparation et ajout des ncAA (semaine 3) :
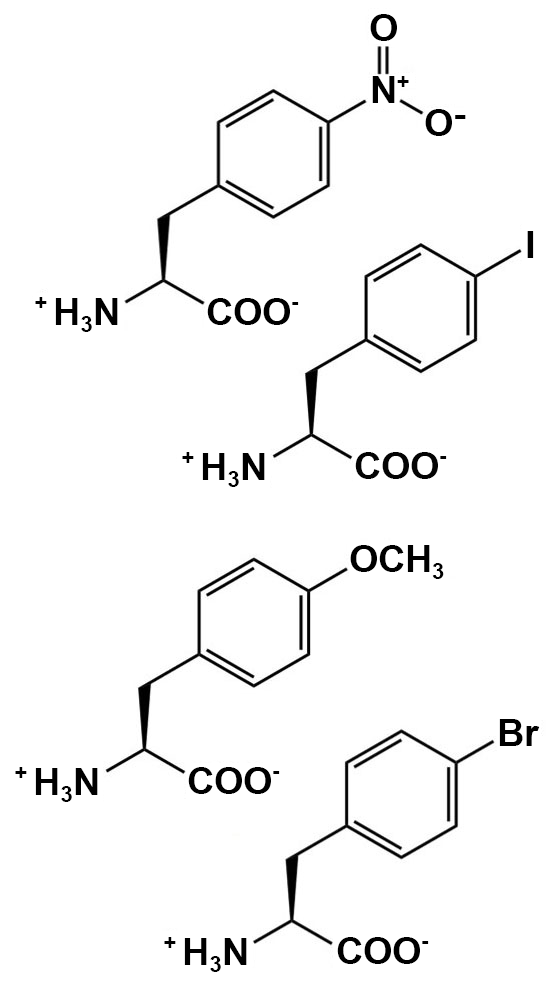
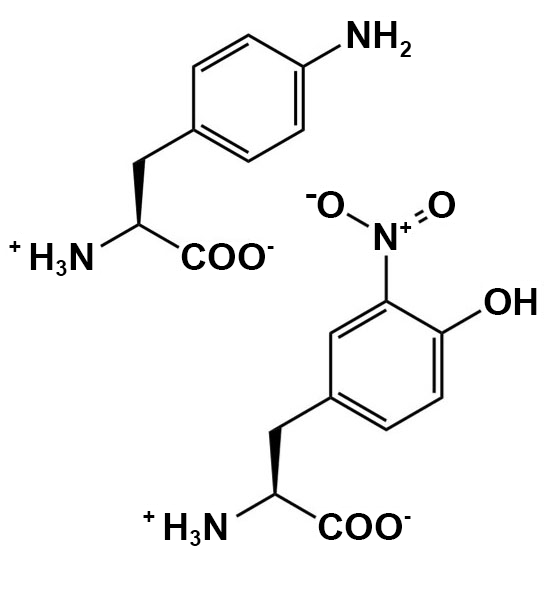
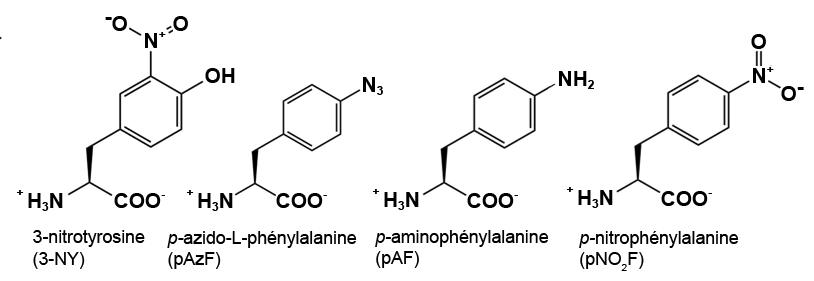
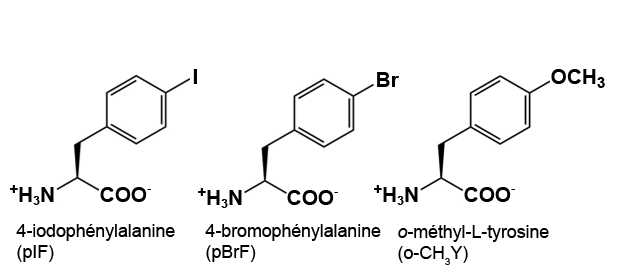
La concentration recommandée pour les ncAA est une concentration finale dans le milieu d’expression > 1 mM. Les ncAA peuvent avoir besoin d’aide pour se dissoudre avant de les ajouter au milieu, selon les caractéristiques chimiques du ncAA. Il est recommandé de peser légèrement plus que la quantité appropriée de ncAA directement dans un microtube à centrifugation. Commencez par ajouter 0,5 ml d’eau stérile et mélangez – si le ncAA se dissout, ajoutez-le à la fiole d’expression appropriée. Si la majeure partie du ncAA se dissout, essayez d’abord d’ajouter 0,5 ml d’eau stérile. Si très peu de ncAA se dissout après le premier 1 ml, essayez d’ajouter 1 équivalent molaire de NaOH à partir de la solution mère à 8 M (ajoutez 5 µl de NaOH à 8 N à la fois et ne dépassez pas 20 µl au total). Trop de NaOH peut endommager certains des ncAA et causer des problèmes de croissance cellulaire. Une fois que les ncAA sont ajoutés à leurs fioles appropriées, laissez-les incuber tout en les agitant à 250-300 tours/minute à 37 °C jusqu’à 48 heures (cette longue incubation est conçue pour respecter l’horaire de BB 494; les cultures sont généralement cultivées pendant 24 à 40 heures avant la récolte).
Pour obtenir un point de temps zéro d’expression protéique, retirez 250 microlitres des cultures cellulaires de départ d’origine en milieu non inductif et centrifugez entre 3 000 et 5 000 rcf pendant 5 à 10 min. Jetez le surnageant et entreposez le petit culot cellulaire à -20 °C dans votre congélateur (étiquetez bien tous les échantillons afin que vous et vos partenaires puissiez bien les identifier à l’avenir).
Exprimez la protéine pendant 24 à 48 heures. Avant de récolter les cellules par centrifugation, n’oubliez pas de retirer d’abord 250 cellules microlitres pour les derniers points de temps pour un gel brut. Déterminez également la densité (OD600) des cultures. Divisez chaque échantillon de cellules en aliquotes de 25 à 35 ml dans des tubes coniques pré-pesés. Faites tourner les cellules dans des tubes coniques de 50 ml pendant 10 min entre 5 000 et 8 000 rcf dans la centrifugeuse de la paillasse (pas la Sorvall). Retirez le surnageant en le versant, déterminez la masse de chaque culot cellulaire et conservez les culots cellulaires à -80 °C. Encore une fois, indiquez bien la section, le numéro de groupe, la date et l’échantillon sur les tubes. Conservez les culots cellulaires dans de petits sacs à fermeture à glissière bien étiquetés au stylo directement sur le sac, indiquant votre section de laboratoire, votre numéro de groupe et la date. N’utilisez pas de ruban adhésif pour étiqueter les tubes ni les sacs : il tombera à -80 °C.
Réflexions approfondies sur les souches bactériennes et les plasmides
Nous vous fournissons des cultures de départ cultivées dans des milieux non inductifs avec lesquels inoculer vos plus grandes cultures, mais qu’utilisez-vous exactement? Prenez-vous aveuglément ce que l’assistant ou l’enseignant vous donne en suivant les instructions pour exprimer les protéines dans ce manuel ou savez-vous comment fonctionne le système et ce que contient exactement cette culture de départ?
Si vous ajoutez des antibiotiques à votre milieu, pourquoi? Comment savoir quels sont les bons antibiotiques? Toutes vos expressions n’utilisent pas les mêmes antibiotiques, pourquoi? Comment se transmet la résistance à ces antibiotiques?
Il existe une grande variété de souches bactériennes différentes que nous pourrions utiliser, chacune présentant des caractéristiques uniques optimisées pour des objectifs précis et présentant des avantages et des inconvénients. Certaines sont conçues pour contrôler étroitement l’expression et sont utilisées pour exprimer des protéines qui peuvent être toxiques pour une cellule. D’autres manquent ou contiennent des mutants de certains gènes afin de prévenir la dégradation des protéines, de favoriser la formation de liaisons disulfure ou de limiter la production de certains métabolites. Quelle souche bactérienne utilisez-vous? Quels sont certains de ses avantages et inconvénients? À votre avis, pourquoi a-t-on choisi cette souche pour ces expériences?
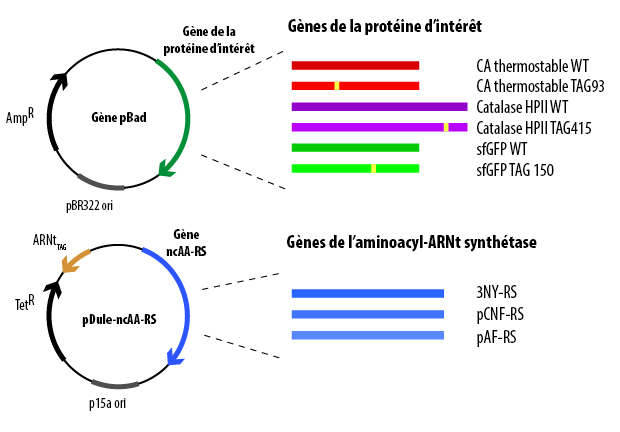
En raison des contraintes de temps de ce cours, les bactéries contenant les plasmides appropriés vous ont également été fournies. Ce n’est pas parce que vous n’avez pas réalisé cette partie de l’expérience vous-même que vous devez ignorer comment cette étape a été réalisée. Comment ces plasmides ont-ils été générés? Que contient chacun des plasmides? Comment ces plasmides sont-ils entrés dans la bactérie où votre protéine d’intérêt peut être surexprimée?
À tout le moins, vous voudrez connaître et comprendre : le type d’E.coli que vous utilisez, les plasmides que vous utilisez et ce qui est codé sur chacun de ces plasmides.
|