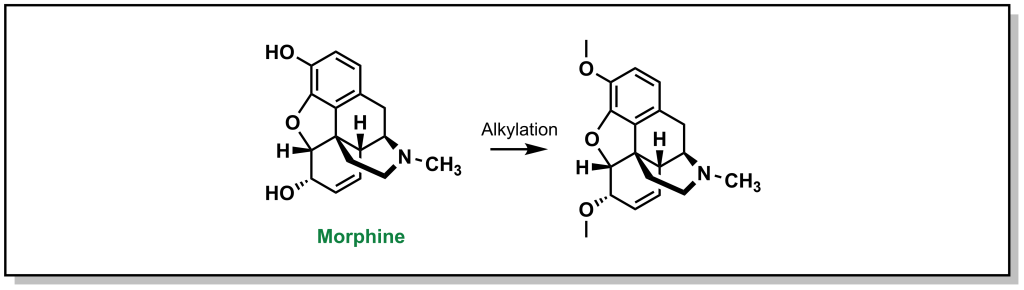
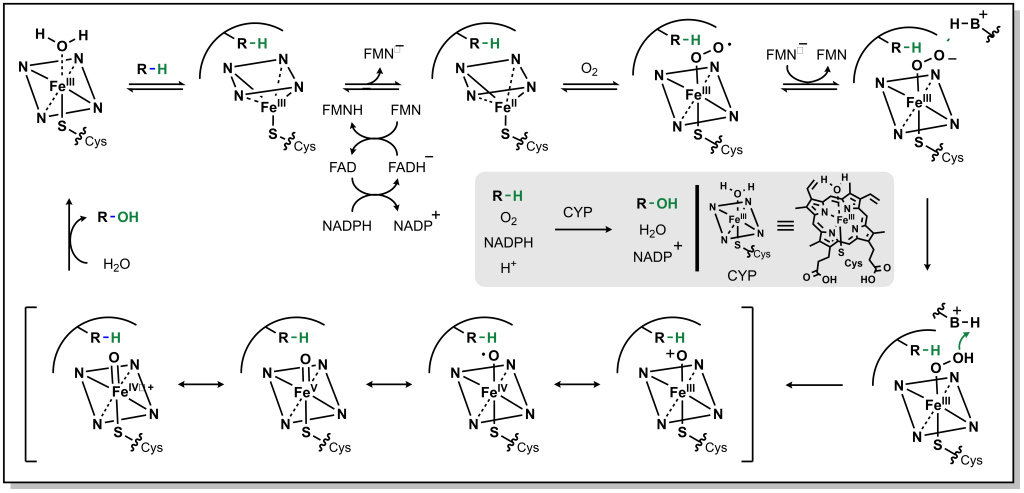
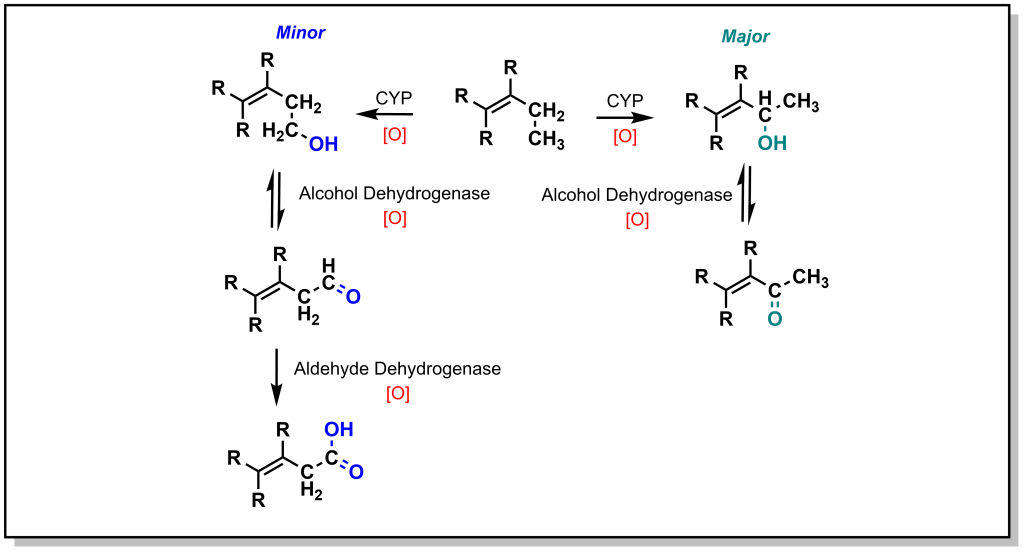
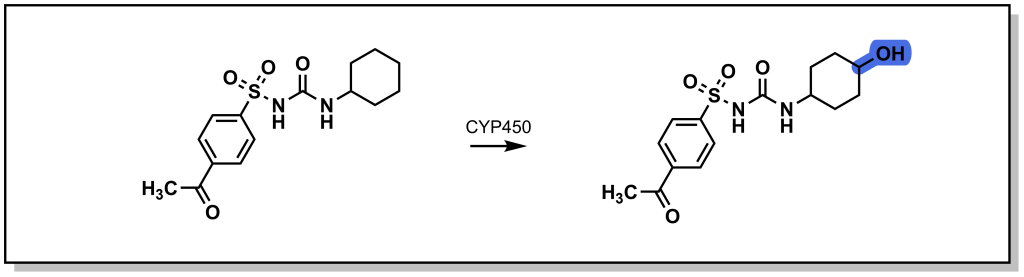
The roots of medicinal chemistry can be traced back nearly a century, to the pioneering work Paul Ehrlich, and his search for a “magic bullet” cure for syphilis infection (caused by Treponema pallidum infection). Over the decades to follow, scientific breakthroughs and technology advancements continued to drive the field of medicinal chemistry, giving rise to the modern omnipresence of pharmaceutical drugs in western society. However, until recently (roughly the turn of the century), drug discovery efforts remained largely centred around a classical pipeline involving linear and iterative research and development approaches. Basic research, focused on the identification and understanding of disease pathologies, was generally a pre-requisite for investigating therapeutic intervention. Selection of a suitable (often reused) target is followed by hit-to-lead studies involving the synthesis of chemical libraries, alongside assay development and execution at the in vitro, cellular, and in vivo stage to provide efficacious compounds. Simultaneous evaluation of the chemical and physical properties (pharmacokinetics) of potential drug candidates is integrated into the discovery pipeline in order to afford desirable properties such as oral bioavailability. Iterative optimization of the molecular structure eventually produces the pre-clinical candidate(s), which are submitted to the Food & Drug Administration (FDA) as well as other regulatory agencies and advanced to clinical trials. Indeed, it is commonplace for drug discovery researchers to boast specific areas of specialty, such as chemical synthesis, pharmacology, molecular biology, or bioanalytical chemistry (to name a few). However, true medicinal chemists are well-versed in all facets of the drug development process, driving the advancement of bioactive molecules to safe, efficacious therapies for patients. This resource is intended to guide the reader through the (small molecule) drug discovery process from a medicinal chemist’s perspective, providing the knowledge and tools required to interrogate the structure and function and bioactive molecules.
How are Drugs Discovered?
2
“The best way to discover a new drug is to start with an old one.”
– James Black
2.1 Natural Products and Natural Product Analogs
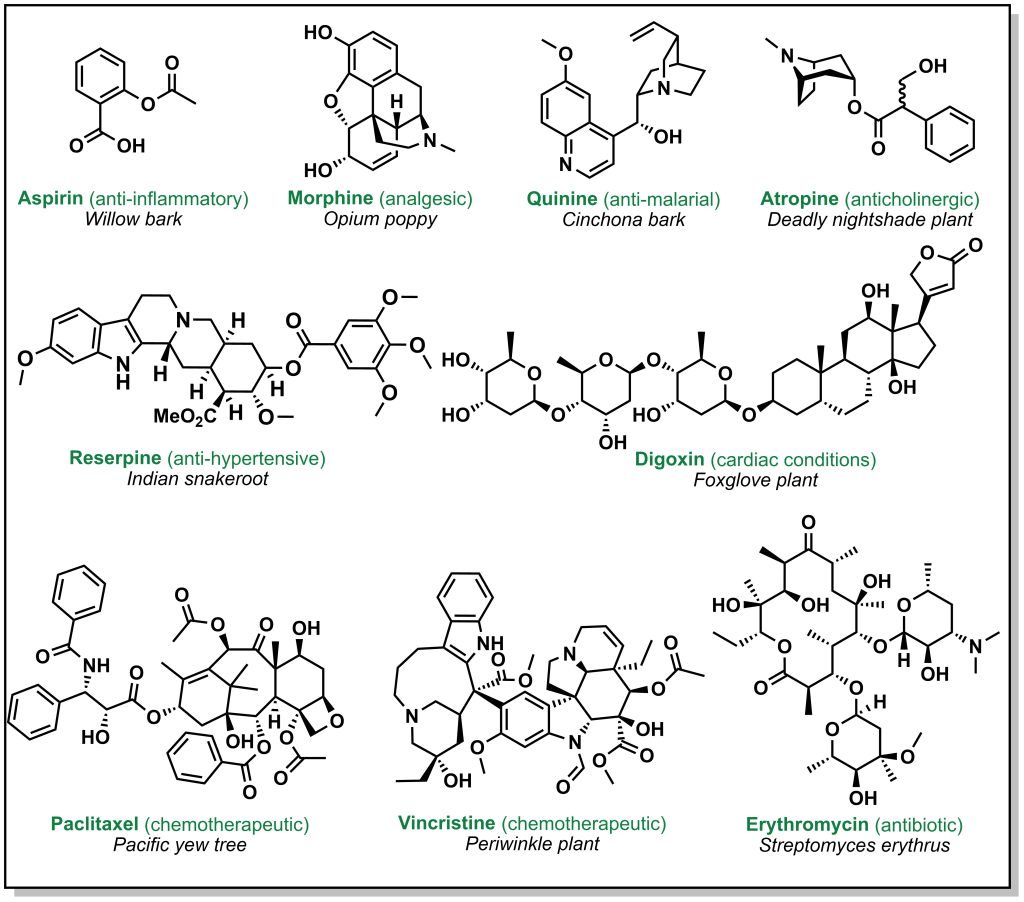
Drug discovery is not always carried out at a molecular level. Herbs, berries, roots, barks, and other natural products have been go-to ailments for humans since antiquity. These natural medicines were often ‘prescribed’ without any prior knowledge of their active constituents. Over time, multiple ancient populations accumulated their own pharmacopeia for natural products, several of which have served as inspiration for the isolation and/or synthesis of more innovative and potent compounds. These remedies derived from plants, animals, or microorganisms have foundational properties that transformed the landscape of modern medicine. Figure 2.1 highlights currently employed drugs that were derived from natural sources along with a selection of case studies below.
FIGURE 2.1 Drugs derived from natural sources.
2.1.1 Aspirin
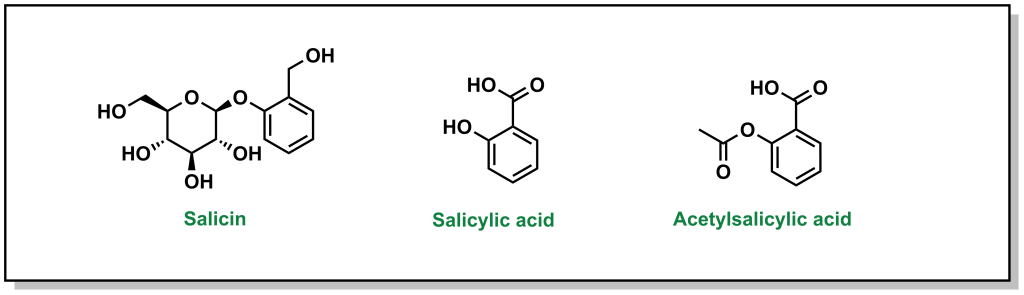
The discovery of aspirin and its growth to one of the most widely used over-the-counter analgesic (“pain-relieving”) drugs, spans multiple millennia, and involves many landmark developments in isolation and synthetic chemistry. Different civilizations independently converged on the use of willow trees, particularly the white willow (Salix alba) and meadowsweet (Filipendula ulmaria), for fever-reducing and pain-relieving properties. The earliest documented use can be found in the Ebers Papyrus, an ancient Egyptian medical text (3000 BC) although Reverend Edward Stone (1763) is often credited with the first scientific study in demonstrating the therapeutic properties . In 1828, Professor Joseph Buncher (Germany) was able to extract the active ingredient (which he named salicin), and it was later refined to salicylic acid by French chemist Charles Gerhardt (1853). Dr. Felix Hoffman (a German chemist at Bayer) was able to acetylate the compound, which alleviated several adverse side effects associated with salicin and salicylic acid, resulting in the compound acetylsalicylic acid (Figure 2.2). This landmark discovery represents the first synthetic drug from pharma, which galvanized a new era in therapeutics. Bayer registered the drug under the name Aspirin in 1899, with the namesake as a derivatization from yet another natural species of meadowsweet (Spirea ulmaria).
FIGURE 2.2 Salicin (active ingredient) and the refined derivatives, salicylic acid and acetylsalicylic acid.
2.1.2 Morphine and Codeine
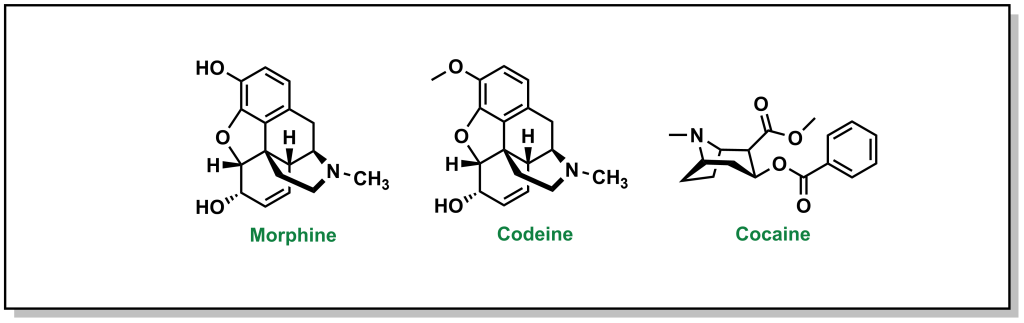
The powerful analgesic properties of the poppy plant (Papaver somniferum) were also known by multiple civilizations, with archaeological evidence of human use as far back as 5000 BC in the Mediterranean region. The seed capsules of poppy plants contain a milky substance called latex that contains opium. Opium is comprised of a mixture of chemical compounds called alkaloids (a loosely defined subset of naturally occurring organic compounds containing at least one nitrogen). In 1804, the German pharmacist, Friedrich Sertürner, isolated morphine (Figure 2.3) from opium (with the name emerging from Morpheus, the Greek god of dreams). Similarly, French chemist Pierre-Jean Robiquet was able to isolate codeine (parent compound of morphine) as another component from opium in 1832. Codeine was named after the Greek word “kodeia” referring to poppy head. Other euphoria-inducing drugs can also be synthesized from opium including heroin and oxycodone, although these are all tightly controlled substances due to their heavily addictive properties.
Figure 2.3 Analgesic compounds morphine, codeine, and cocaine.
2.1.3 Penicillin
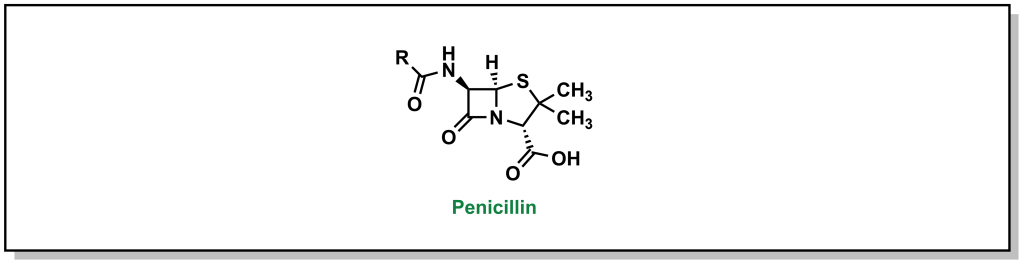
In 1928, Scottish physician, Alexander Fleming, was conducting research on the growth properties of the bacteria, Staphylococcus aureus. He returned from a two-week vacation and discovered that his cultures were contaminated by mold, but also that the bacteria could not grow in close proximity to the fungi on the culture plate. The fungi was determined to be Penicillium notatum. After culturing it, Fleming confirmed that the growing broth harboured an anti-bacterial substance that was effective on Gram-positive pathogens but not on Gram-negative bacteria. In 1929, he re-named the broth, penicillin, in place of the more colloquial name, “mold juice”. The chemical structure was determined and purified by another team (Howard Florey and Edward Chain) in 1940, followed by the first treatment in patients in 1942. (Figure 2.4) Its uptake into the clinics was largely accelerated during World War II with its success in tackling bacterial infections and the trio of scientists awarded the Nobel Prize in Physiology or Medicine in 1945.
FIGURE 2.4 Structure of penicillin.
2.2 Modern Rational Drug Design
One of the common themes evident in drug discovery via natural products is that it is grounded in astute observation of often-serendipitous circumstances, followed by empirical trial-and-error methods, and ultimately isolation of the key active ingredient. Over the early 20th century, this framework for drug discovery transitioned to a more step-wise hypothesis-driven approach with the goal of systematically developing, synthesizing, and exploring new (non-natural) composition-of-matter. This reform in therapeutic methodology was accelerated by the creation of the US FDA in 1938, followed by substantial administrative changes in the Kefauver-Harris Drug Amendments of 1962, as a result of the thalidomide drug scandals. The US FDA oversees public health in the US including the approval of new drugs and medical devices. The Canadian counterpart is referred to as Health Canada, which was instituted in 1919 in response to the Spanish Flu outbreak, and later reformed in 1993. Both Health Canada and the US FDA collaborate closely (via the Regulatory Cooperation Council Joint Forward Plan) to harmonize several drug-related policies, although they are separate entities and can/will uphold different guidelines.
2.3 Typical Stages of the Drug Discovery Pipeline
For a medicinal chemist, the stages of the drug discovery pipeline are often represented in a linear, forward-moving strategy, although the overall process is highly iterative and can involve multiple cycles of optimization and re-visiting of each stage (Figure 2.5).
FIGURE 2.5 A general drug discovery pipeline schematic.
2.3.1 Stage 1: Target Selection
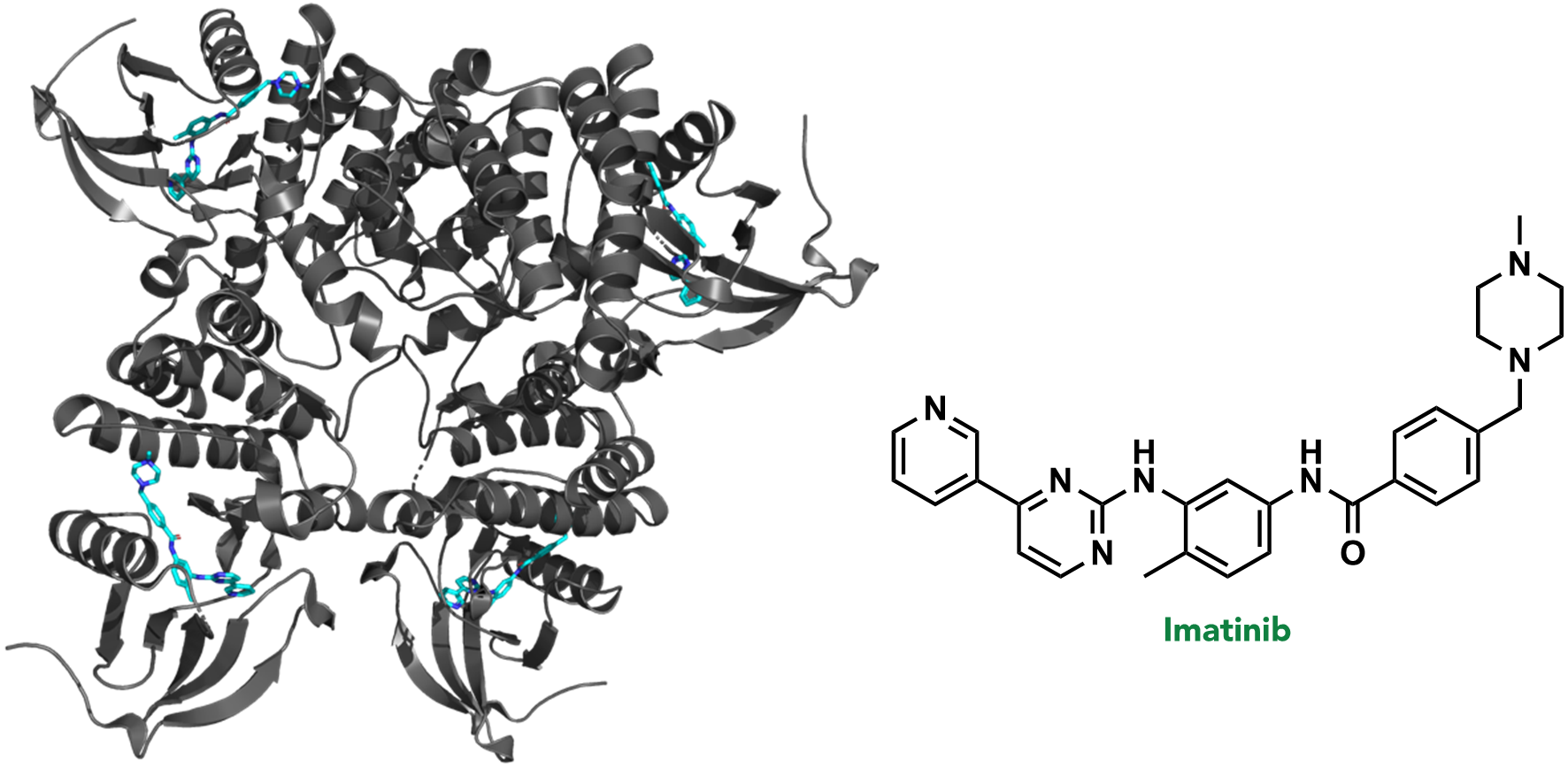
In the first stage of drug discovery, a target or specific disease is selected. A target refers to a biomolecule where pharmacological or genetic intervention would (directly or indirectly) disrupt a critical pathological biochemical process. For example, >95% of chronic myeloid leukemia (CML) cases result from a random genetic mutation that creates the fusion protein, BCR-ABL kinase. BCR-ABL kinase is therefore a target in CML, and is often treated with the drug, imatinib (or one of its analogues). (Figure 2.6)
One disease can have more than one target, and similarly, one target may be relevant in more than one disease. The rationale for selecting a target is usually supported by an array of genetic, biological, and physiological data that suggest it will be efficacious to knock-down while not compromising overall safety to the patient. This is generally referred to as target validation. For example, disrupting the enzyme Protein Kinase A (PKA) is not a feasible option in the rare fibrolamellar hepatocellular (FLC), despite the fact that its variants have been shown to drive this cancer. This is because of the importance of PKA in the heart for maintaining appropriate cardiac functioning. However, there are many cases where background biological information is not readily available, or the target is not well-studied. In these cases, a greater importance is placed on pre-clinical investigations and safety trials.
FIGURE 2.6 Imatinib bound to ABL kinase (PDB: 2HYY).
2.3.2 Stage 2: Hit Identification and Hit-to-Lead / Lead Discovery
Following selection of the disease/target, the next stage involves identifying a chemical compound that can interact with the target. Usually, a series of compounds are evaluated in an experiment that involves the desired target. This experiment, called an assay, can involve a variety of tools to quantify the interaction between the compound and biomolecular target; for example, monitoring the interaction (binding event) via nuclear magnetic resonance (NMR), quantifying a change in the target activity via fluorescence, or observing cell death via imaging. The types of assays will vary, depending on both the target and the nature/origin of the compounds being tested, and are often custom-designed for each drug discovery program.
The output of the assay will usually provide an initial group of compounds that can engage with the target, which are referred to as hit compounds. The activity of these hit compounds can vary substantially, but they serve as general starting points to create additional generations of molecules with better activity. The goal of subsequent organic synthesis campaigns is to improve the hit compounds and create/select a lead compound. A lead compound is a compound that:
Interacts with the target to achieve the desired biological activity.
Is amenable to synthetic modifications.
Can reach the target once administered.
Distinguishing and selecting lead compounds will be discussed later in this text. However, an initial hit molecule differs from a final lead molecule in that it usually has lower biological activity, and has not been validated to reach the target upon administration. As such, these two properties are classical outcomes of hit-to-lead or lead discovery campaigns, where the goal is to chemically synthesize new molecules that build and optimize a hit compound into a lead molecule.
2.3.3 Stage 3: Lead Optimization
Although identifying a lead molecule is a monumental step forward in the drug discovery pipeline, it is often chemically distant from the final drug candidate. This is usually because the lead molecule has specific properties that can limit its efficacy. For example, the structure-activity relationship (SAR) optimization rounds from the previous hit-to-lead studies may have identified specific functional groups required for target binding (biological activity), but the oral absorption of the compound may not be sufficient, as a result of these chemical groups. Therefore, at the lead optimization stage, the goal is often to preserve the structural elements that maintain potency of the lead compound, while optimizing a new molecule with improved physical properties for bioavailability. It is conceivable that potency of the newly developed candidate molecules may be reduced, as other parameters are optimized. This is a balancing act to fine-tune the optimal overall properties between potency, efficacy, permeability, and stability.
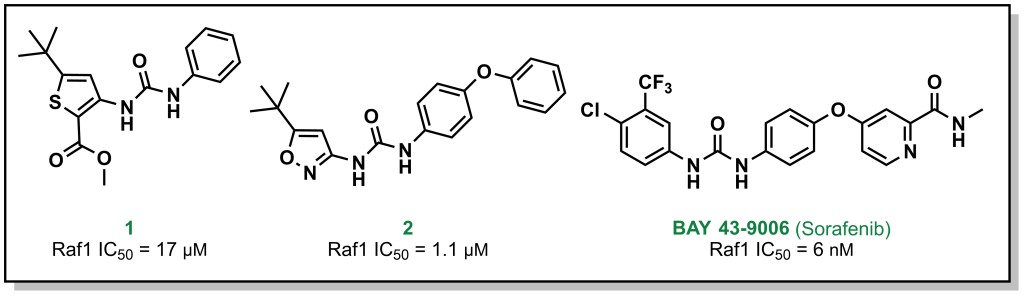
For example, Bayer (in partnership with Onyx Pharmaceuticals) initially screened ~200,000 compounds to explore the target Raf1 kinase, which led to the initial hit molecule 1 (Figure 2.7) with a half-maximal inhibitory concentration (IC50) of 17 µM. The urea and phenyl group were identified to be critical for potency and another ~1000 bis-aryl urea analogs were generated that led to an isoxazole derivative as the lead compound (2) (Figure 2.7). The compound underwent lead optimization to the final development candidate BAY 43-9006 (sorafenib) with a potent affinity for Raf-1 (IC50 = 6 nM). Crystallographic studies (which were determined retrospectively) revealed that the urea moiety was crucial in the SAR, due to the formation of two critical H-bonds (with the backbone aspartic acid residue of the DFG loop and the glutamate side chain of the αC helix of the target protein – structural elements that will be discussed in Section 2.5.1). Additional key features from BAY-43-9006 that were identified include the 4-pyridyl ring that mimics the natural adenine scaffold and the chlorobenzene ring which interacts with a hydrophobic pocket behind the orthosteric ATP binding site. Although the lack of structural information did not allow for visualization of these effects at the time of compound synthesis, these features improved the binding capacity of the drug which was represented by the lower IC50 values and indicated that they were important in the lead optimization process.
FIGURE 2.7 Representative examples of the SAR campaign that led to sorafenib targeting Raf1 kinase.
2.3.4 Stage 4: Preclinical / Clinical Development & Regulatory Approval
The focus of this resource is on medicinal chemistry, and following completion of lead optimization and pre-clinical trials, the compound is generally optimized and there is a diminished motivation for synthesizing newer compounds. However, the subsequent regulatory steps are briefly summarized below.
Following lead optimization, the top developmental candidate needs to be evaluated in more complex models. This usually involves a mammalian model such as a mouse or rat that has been modified to express a phenotype representing the disease state. For example, exploring cancer tumors can involve engrafting specific cancer cells into a mouse and monitoring the tumor volume upon compound treatment (as a measure of cancer-killing potential). For neurological diseases, this could involve genetically introducing known mutations and monitoring specific exercises such as novel object recognition time tests (as a measure of memory function). Generally, these studies are benchmarked to the standard-of-care which is the current drug regimen used in clinical settings, and this provides an understanding of the degree to which the candidate molecule can improve outcomes. Although it is not an absolute requirement, usually two preclinical models are required to advance to human trials.
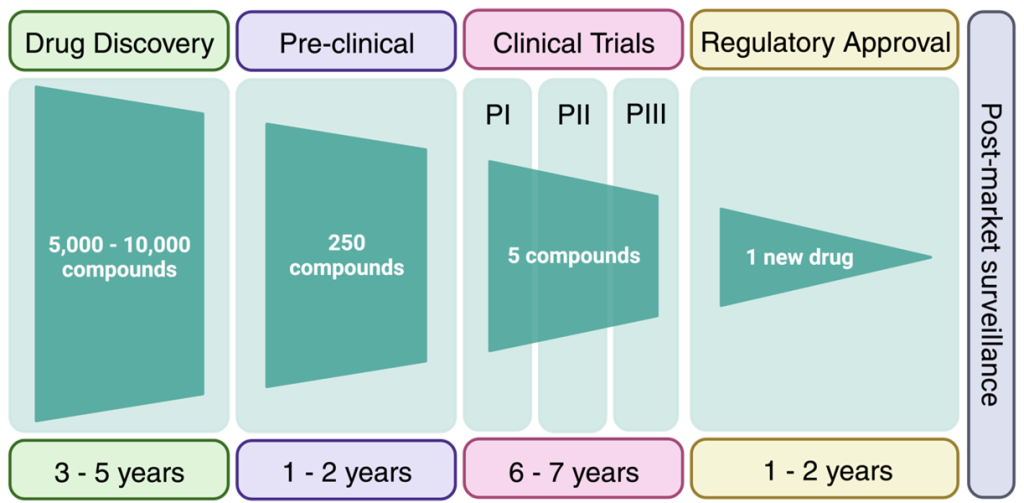
Clinical trials are an expensive and complex endeavour with the goal of demonstrating that the drug candidate is safe and efficacious. Biotech companies looking to evaluate their drug candidates in humans (in Canada) require approval from Health Canada. The classical route involves Phase I trials, which are predominantly focussed on safety and pharmacokinetics of the drug candidate in healthy individuals. However, for rare diseases, the compound may be administered to a population with the disease of interest in Phase I. Following positive results, a Phase II clinical trial would be initiated with a cohort of patients from the disease population. Phase III clinical trials would have an even larger disease population for improved sampling statistics. Depending on the type of dosing performed, the clinical trial may also be labelled with an a or b suffix. Clinical trials cost millions of dollars and require years to prepare clinical centres and teams, recruit patients,
FIGURE 2.7 Timeline for advancing a drug candidate to the market. (Image adapted from Matthews et al. Proteomes 2016)
administer the drugs, and analyze the results. On top of this, the success rate of a drug to advance through all three clinical trials with positive data is ~7%. Overall, it is estimated that advancing a compound through the drug discovery pipeline may take 15-20 years and cost 1-2 billion dollars. Although this may seem daunting, understanding why drugs fail at the late stage, helps front-load these issues into the medicinal chemistry thought process at the beginning of the drug discovery pipeline. The results of many clinical case-studies have revealed benefits and challenges of different functional groups and the goal of medicinal chemistry is to build on these learnings to avoid attrition of drugs at late clinical stages.
2.4 Rational Drug Design: The “Magic Bullet” Concept
Paul Ehrlich, a German physician, pioneered a number of concepts that took a foothold in the early applications of medicinal chemistry. One of his primary contributions was in the field of immunity, for which he was awarded the Nobel Prize in Physiology or Medicine in 1908. He developed a “side-chain theory” which purported that specific chemical structures could elicit a response by immune cells in the blood, similar to the protein-ligand “lock-and-key” biochemistry model. He also adopted the concept of a “magic bullet” (a concept from German stories of a bullet that locks on to a target and cannot miss once fired), proposing that it would be possible to develop a compound that mimics these properties and specifically engage invading foreign entities with a high degree of specificity.
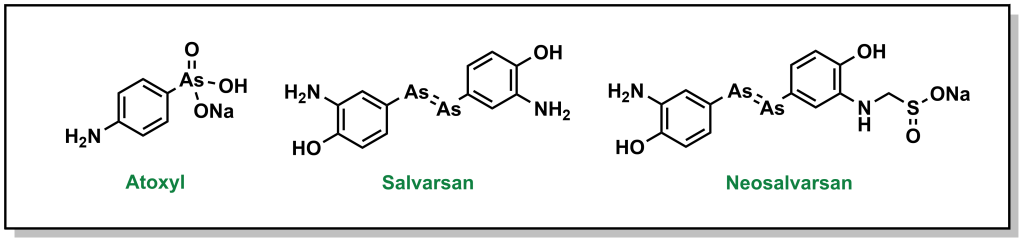
Ehrlich pursued his idea of a magic bullet, generating libraries of compounds based on the toxic drug atoxyl for the treatment of syphilis, which was a major public health threat in Europe at the time. His goal was to identify a compound that would selectively kill the responsible bacterium, Treponema pallidum, without harming healthy cells. Eventually, his team developed compound 606 or arsphenamine which was marketed to Hoechst AG under the name salvarsan in 1910. Although this compound was a substantial improvement over the standard-of-care (mercury-containing compounds), it was highly unstable in air, which could lead to multiple and serious adverse side effects. Another 300 derivatives later, his team developed a more degradation-resistant drug, which was called neosalvarsan (Figure 2.8). Although these compounds were superseded by the discovery of penicillin in the 1940s, the approach adopted by Ehrlich foreshadowed the iterative drug discovery pipeline to optimize new selective compounds.
FIGURE 2.8 Structure of atoxyl and its derivatives.
2.5 Rational Drug Design: Understanding the Target
Understanding the target will shape the drug discovery program and alter the overall medicinal chemistry strategy. If considering just the human genome, there are approximately 20,000 genes that encode potential targets. About 15% of the genome can be pharmacologically altered with today’s collection of drugs. This means that there are potentially thousands of targets without drugs (although not all of these proteins are disease-relevant or would be significant effectors of disease if targeted by a drug). Additionally, there are a wealth of non-human proteins, such as pathogenic biomolecules from bacteria or viruses that cannot be accessed by current drugs.
Although there are many different pathways, and therefore many protein targets that require pharmacologic intervention, current targets are asymmetrically skewed toward certain privileged protein families. For example, nearly two-thirds of all drugs on the market, target proteins from either the kinase or G-protein-coupled receptor (GPCR) superfamilies. Part of the reason why these protein classes are over-represented in drug discovery is because they exist at the top of large biochemical cascades, and therefore blockading their action can yield powerful responses. GPCRs and nuclear receptors have many different types of drugs that act on the same target (i.e. there are many selective drugs for these targets), whereas for kinases the reverse trend is true – there are many different kinases that are targeted by a smaller number of drugs, which indicates many more “off-targets”. Additionally, GPCRs are located on the cell surface, which reduces the requirement of a drug to cross an additional membrane. Importantly, kinases have been heavily studied both in structure and function which helps guide the drug discovery process.
Designing drugs based on protein structure represents a critical step in the rational development of drugs based on protein-ligand interactions. Although the “lock-and-key” model of protein-ligand binding has been updated to involve “induced-fit” models that account for the fluidity of the protein and ligand conformations, it still provides a useful classical analogy for understanding the goals of designing new drugs for protein targets. This model has been applied to understand the different conformations of kinases and how drugs can engage these targets.
2.5.1 Kinases
Protein kinases are enzymes that are responsible for transferring a phosphate group from ATP to another protein with a hydroxy-side chain (e.g. tyrosine). This post-translational modification introduces a di-anionic charge which is unique to a phosphorylated residue (and not found on any naturally incorporated amino acid). As such the consequences of phosphorylation can result in drastically altered protein conformations or interaction partners. There are two main types of kinases, Serine/Threonine kinases (~385 proteins) and Tyrosine Kinases (90 proteins) which specifically phosphorylate their namesake residue. Phosphorylation via Ser/Thr kinases is usually associated with large-scale conformational changes whereas as action by Tyr kinases can result in protein localization changes. There are also rarer kinases that are capable of phosphorylating non-conventional residues such as Histidine, Arginine, and Lysine.
The kinase structure is highly conserved and the secondary structural elements are well-defined. The active kinase domain contains two lobes, a small N-lobe (largely comprised anti-parallel β-sheets) and a large C-lobe (largely α-helical) connected by a hinge region. The ATP binding site is sandwiched at the interface of these lobes and the adenine ring forms H-bonds with the hinge region.
Kinases cycle between an inactive (open) conformation and an active (closed) conformation. The open conformation enables ATP / ADP to access the active site hinge region, whereas the closed conformation facilitates formation of the functionally active cleft. Both the N- and C- lobes have key structural features associated with each state. The N-lobe contains a large helix (labelled an αC-helix) that forms the roof of the active site and will either rotate away (open/inactive) or towards (closed/active) the orthosteric site. The C-lobe contains a conserved loop (referred to as an Activation or DFG loop) that begins with residues Asp, Phe, and Gly and ends with Ala, Pro, and Glu. The Asp side chains bind Mg2+ which helps coordinate the phosphate of ATP. In the active conformation, the Asp faces into the pocket, whereas the inactive conformation has the Asp extruding out of the pocket. (These conformational switches also have the Phe entering and blocking the adenine binding pocket).
Kinases represent the archetypical drug binding sites – they contain a well-defined binding pocket that is buried, hydrophobic, with affinity for a known ligand (ATP). In fact, the binding pockets of kinases and the inhibitors that have emerged are so extensively studied that they are stratified across 6 types:
Type I inhibitors bind directly at the orthosteric (ATP binding) site and leverage conserved catalytic residues in their interactions. These inhibitors are ATP-competitive and have excellent shape complementary by exploiting the rigid nature of the active site. For example, crizotinib and dasatinib are Type I kinase inhibitors.
Type II inhibitors bind at the ATP binding site but in the DFG-out (inactive) conformation. In this conformation, a new hydrophobic site is exposed between the C-helix of the small lobe and the DFG motif. The example of sorafentib, which is discussed above, is a Type II inhibitor.
Type III inhibitors bind deeper in the ATP binding site, in a hydrophobic pocket behind the active site. These inhibitors have substantially improved selectivity, since this hydrophobic pocket is kinase-specific. Moreover, these inhibitors do not have a heterocyclic group that normally mimics the adenine ring. However, this pocket is often transient and occupancy does not guarantee inhibition.
Type IV inhibitors bind at an allosteric site, completely distinct from the ATP site.
Type V inhibitors are bivalent inhibitors with chemical moieties that engage with structural elements on both lobes of the kinase.
Type VI inhibitors are covalent inhibitors that will form a covalent bond with a residue (usually cysteine) on the kinase.
An important aspect for inhibitors is that over time, cells can become resistant to the effects of inhibitors. This can emerge from mutations in the target that reduce binding affinity or capacity of the inhibitor. In kinases, this can often occur with a gatekeeper residue, which is a key residue that is located close to the hinge region and guards access to the pockets behind the adenine ring. In BCR-ABL1 (CML) the T315I gatekeeper mutation results in steric hindrance that impedes binding of imatinib. In EGRR the T790M gatekeeper mutation induces resistance by increasing affinity for ATP. New drugs need to be designed that can overcome these mutations by avoiding the gatekeeper, allosterically engaging the target, or harnessing a separate biochemical pathway altogether. For example, the drug ponatinib bypasses the gatekeeper mutation via an ethynyl group, whose linear structure sterically evades the bulky T315I mutation and can continue to block BCR-ABL activity.
What are Properties of Hit and Lead Compounds?
3
“If you do not expect to, you will not discover the unexpected.”
– Heraclitus
3.1 Sources of a Lead compound
We have previously defined important properties for a lead compound. A lead compound is a compound that:
Interacts with the target to achieve the desired biological activity.
Is amenable to synthetic modifications.
Can reach the target once administered.
Although a significant amount of time and resources are usually invested into identifying a lead compound, they usually serve as starting points for large scale chemical optimization to evolve into safe and effective drug candidates. Lead compounds can arise from different sources as described below.
3.1.1 Natural Extracts
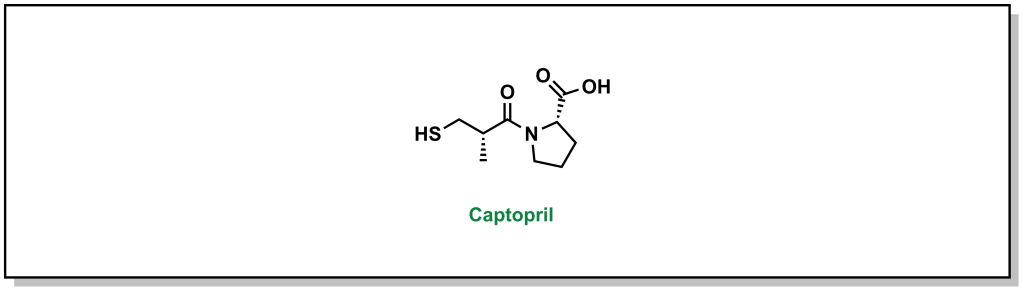
Many lead compounds are derived from plants, animals, or microorganisms as seen in Chapter 2. Roughly 35% of all current medicines originated from natural sources. Natural products are known for having unique chemical structures and broad diversity in composition of matter. These compounds often contain stereogenic modalities and involve high complexity that translates into diverse biological activity. These natural extracts may contain mixtures of hundreds or thousands of compounds and there can be challenges in identifying and isolating the active ingredient from the mixture, as well as developing a viable route to access the compound by chemical synthesis. Natural compounds may also be starting points for further synthetic chemistry screening. For example, captopril is an antihypertensive agent that was identified following studies of the venom of the Brazilian snake, Bothrops jararaca, which identified 9 peptides that potentiated the effects of bradykinin (a naturally occurring peptide that facilitates vasodilation). Bristol Myers Squibb (BMS) used these peptides as templates for synthetic campaigns that eventually led to the drug, captopril (Figure 3.1).
FIGURE 3.1 Structure of captopril.
3.1.2 Prior Art
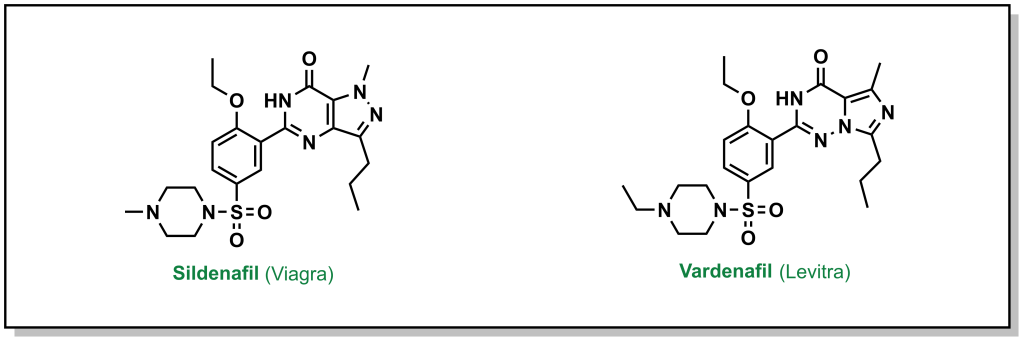
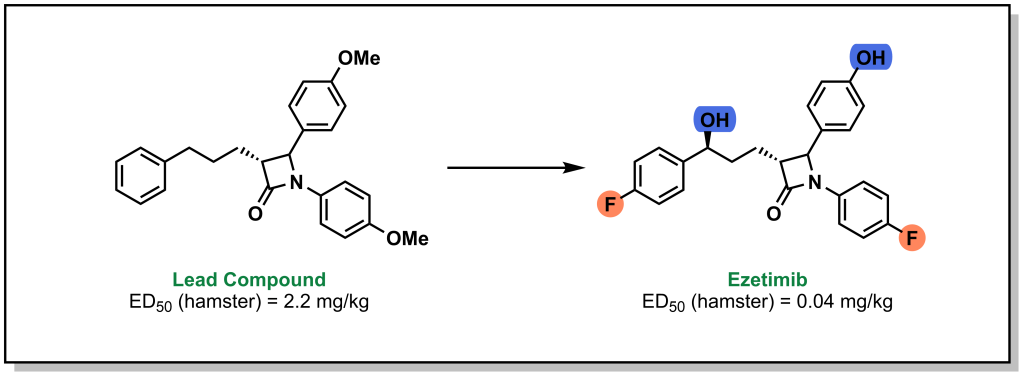
Often lead compounds can be created from modifications of existing drugs. The term ‘prior art’ is borrowed from patent law and refers to compounds that are already known or available in the public domain. This can be convenient because these compounds have safety profiles, synthetic feasibility, efficacy, and metabolic liabilities that are generally understood. As such, prior art can form large aspects of new drug discovery programs. One challenge with prior art is ensuring that there will be ‘freedom-to-operate’ for the chemist on any new composition of matter that would be created, and that the potential new drug could be protected under a new patent. Any derivatives not covered by one patent represent analogues that may be explored by other researchers. For example, consider the patent that covered sildenafil (Pfizer, 1998) and the alternative analogue verdenafil (GSK/Bayer, 2003). Both drugs are currently sold for the treatment of male erectile dysfunction as Viagra and Levitt, respectively (Figure 3.2).
FIGURE 3.2 Structure of sildenafil and vardenafil.
3.1.3 In Silico Screening
With the advent of more powerful computational prediction software in the past 10-20 years, in silico screening has become a mainstream approach for drug discovery. The most routine form of in silico screening involves molecular docking, where the ligand/target interactions of a prospective drug molecule are modelled at the predicted binding site with a three-dimensional conformation of the target. The potential interactions are computationally predicted, optimized, and the compounds are ranked according to parameters such as the free energy of binding (ΔGbind). Thousands of compound libraries can be screened in silico at much higher throughputs and lower resource burden than experimental screens. However, there are limitations that are defined by the structural inputs of the target and the ligand, as well as conformational mobility, especially of the protein target. Many ligands that are computationally predicted from random library screening to bind to the target may not demonstrate any binding, experimentally (in vitro). Notably, there are substantial improvements in predictions if a previous protein-drug structure has been defined (eg. via X-ray crystallography). There are rapidly expanding toolkits of different software packages available for in silico screening; however, any identified hits still need to be experimentally validated. In silico screening is expected to become even more prevalent as computational and AI-driven strategies become more robust, and every steady-state drug screening pipeline will likely involve computational screening at different levels.
3.1.4 Random and Targeted Library Screening
Random screening can involve exploring large chemical libraries without discretion for chemotype or molecule diversity. Since identification of positive hits can rely on serendipity, this is usually performed when there is limited knowledge on the target of interest. The activities of streptomycin and tetracycline were identified in this manner. By contrast, targeted screening can involve a range of chemical moieties and scaffolds that are usually related by at least one chemotype. For example, kinase-targeting libraries can employ molecular species with adenosine as a backbone, and SH2 domain-targeting libraries may focus on short peptides containing a phospho-tyrosine residue. The well understood binding modes of previous inhibitors with these bindings sites provides the chemist with a starting point for building a library of targeted molecules that is more likely to provide positive hit molecules, given the inclusion of an established binding fragment. The scaffolds are often modified with an assortment of chemical moieties and functional groups, which could have a variety of desired impacts such as achieving target selectivity. These types of libraries help improve the probability of obtaining a hit molecule, while also maintaining diversity in the composition-of-matter. One downfall of this approach, relative to random library screening, is the low likelihood of identifying novel binding sites for these protein classes.
3.1.5 Fragment-based Screening
One of the most widely employed strategies in hit-to-lead development is focused on fragment screening libraries. Fragments are low-molecular weight species that are usually characterized by the Rule of Three (the following rules involve parameters that are multiples of 3):
1) The molecular weight of a fragment molecule is < 300 Da
2) The number of hydrogen bond donors ≤ 3
3) The number of hydrogen bond acceptors ≤ 3
4) The number of rotatable bonds ≤ 3
5) The cLogP ~ 3; This parameter refers to the distribution of the compound in a mixture of hydrophobic (octanol) and hydrophilic (water) solvents, and can be experimentally determined in a shake-flask experiment and calculated by the equation: P = [cmpd]octanol / [cmpd]water (1-Octanol is typically used as it is a long saturated alkyl chain with a hydroxy group that mimics the lipid membrane and is sparingly soluble in water). The term cLogD is used to describe cLogP at a specific pH if the compound has an ionization site(s).
Screening libraries of fragment molecules can provide initial hit compounds that will be starting points for eventual lead discovery. However, since these are low molecular weight molecules, they are limited in their capacity to engage in multiple binding interactions and usually need to be modified substantially. Fragments can be altered into lead molecules through different chemical strategies, predominantly through four approaches that were summarized by Rees et al. in 2004 (Nature Reviews Drug Discovery).
3.1.5.1 Fragment Evolution
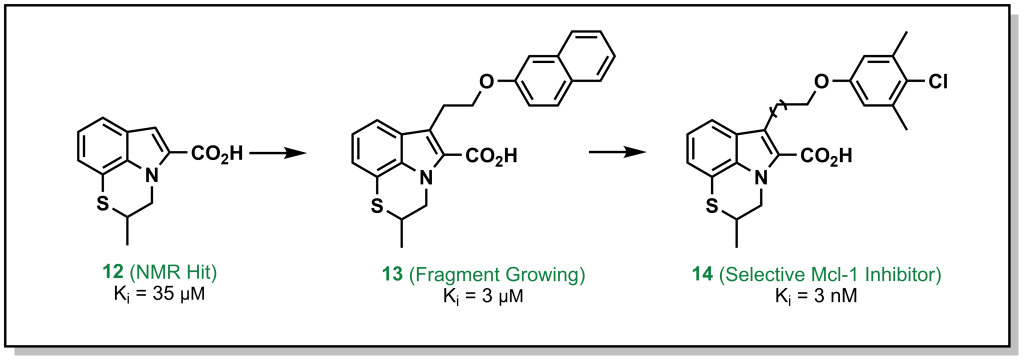
In this approach, a fragment is identified that binds to a specific site on the target, and structural information is employed to build out the fragment and reach other interactions within the binding site. For example, the tricyclic indole 12 (Figure 3.3) derivative was identified from an NMR screen as a modest hit against the anti-apoptotic protein, Mcl-1. Evolution of the hit through accessing hydrophobic interactions led to 10,000-fold improvement in potency over different iterations and introducing functionality to engage with adjacent regions of the active site.
FIGURE 3.3 Fragment evolution of indole derivatives targeting Mcl-1.
3.1.5.2 Fragment Linking
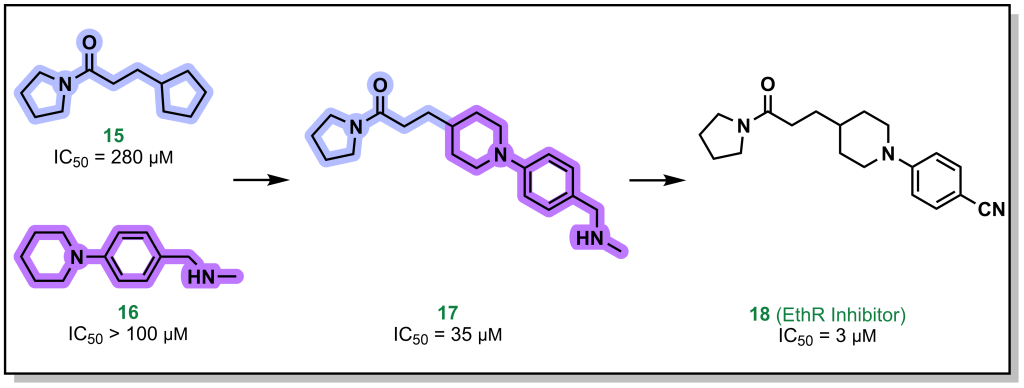
Fragment linking, involves two fragments that bind in close proximity to each other on the target, and can be linked synthetically to provides a higher affinity molecule. For example, the pyrrolidine and piperidine-based fragments 15 and 16 (Figure 3.4) were identified to bind to the protein EthR with high micromolar affinity. Linking both of these proximally binding fragments resulted in 10-fold improvement in affinity which was further improved (another 10-fold) via additional fragment evolution. In some cases, flexible alkyl (or other) linkers are included to idealize distances for binding of both fragments and later optimized in the SAR.
FIGURE 3.4 Linking of fragments targeting EthR improve binding affinity compared to the individual species.
3.1.5.3 Fragment Self-Assembly
Fragment self-assembly is a variation of fragment linking where the fragments can be engineered to possess complementary reactive functional groups. In this way, if the fragments bind in close proximity to each other, they can self-anneal to form a new molecule that has increased potency than the individual fragments A and B (Figure 3.5). For example, the individual fragments have >1 mM binding affinity for CDK2 but following imine condensation form a molecule with an IC50 of 30 nM.
FIGURE 3.5 Fragment self-assembly of molecules in close proximity improves overall binding affinity.
3.1.5.4 Fragment Optimization
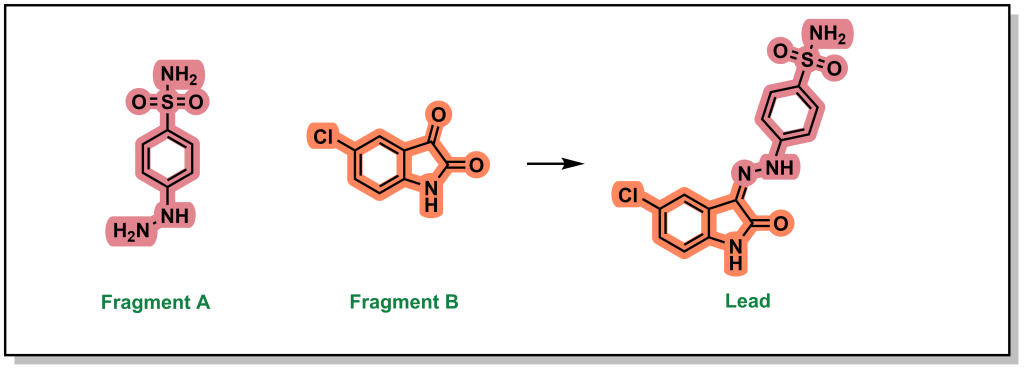
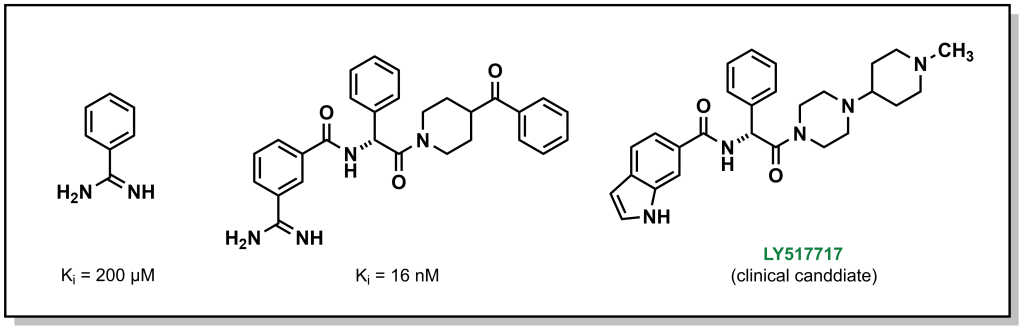
The final approach (fragment optimization) is similar to conventional SAR optimization (i.e. a functional group determined to be relevant for target binding and is modified to increase binding or other desirable molecular properties). For example, drug candidate LY517717 for the blood clotting factor Xa, was built from naked benzimidamide fragments (with >200 µM potency), followed by more advanced in silico screening to design and synthesize larger molecules for bio-assay testing. The original fragment was ultimately replaced to improve oral bioavailability, highlighting the iterative steps involved in building and optimizing a fragment hit into a lead molecule.
FIGURE 3.6 Fragment optimization of the initial scaffold to leverage sites for interaction interfaces.
In all cases, these fragment-based approaches enable synthesizing de novo compounds with the goal of precision-based medicines by elaborating on chemical hits and fragments that have positive binding properties.
3.2 Measuring Target-Compound Binding
A key element of drug discovery programs involves stratifying compounds into potent hit and lead molecules which requires quantifying their capacity to engage with the target to objectively discriminate between compounds. This is most commonly performed through two parameters: i) determining the concentration where a molecule elicits half of its maximal inhibitory effect (IC50) or ii) a dissociation constant (Kd).
IC50 values are one of the most commonly measured parameters in drug discovery. In an assay to determine IC50 , a parameter such as enzyme reaction rate is monitored as a function of the inhibitor/drug concentration (the enzyme and substrate are held at constant concentrations in the assay). The assay is performed multiple times with different concentrations of the inhibitor. In experimental trials where the inhibitor concentration is high, the inhibitor can compete with the substrate to reduce enzyme activity. At a certain inhibitor concentration, 50% of the enzyme activity will be inhibited, which provides the IC50 value (in less common assays, comparable values such as IC30 or IC80 are derived which reports 30% and 80% inhibition respectively). A compound with a lower IC50 value indicates a more potent compound, since it requires a lower concentration to inhibit the enzyme. An important aspect of determining IC50 values, is that if the assay is repeated with increased substrate or more enzyme, the IC50 value will increase (as this requires more drug to inhibit the enzyme). As such, changing conditions for the assay can mis-represent the apparent potency of a drug, and these are relative measurements that can only be compared between compounds if uniform conditions were used across the entire series.
Contrastingly, Kd characterizes the protein-ligand interaction with an absolute value that is intrinsic of the inhibitor and not dependent on enzyme/substrate concentration. In this case, the inhibitor is introduced to the enzyme at different concentrations, and the binding is measured directly (as opposed to another parameter such as reaction rate or product formation). Kd measures the binding equilibrium between the enzyme and inhibitor, and similar to IC50 values, the lower the Kd the more potent the inhibitor. Unlike IC50 values, Kd values for different compounds can be compared and ranked (even in different assay conditions) and a compound with a Kd in the nM regime is more potent than a compound in the µM regime. Hit compounds generally exhibit low to modest µM Kd values whereas lead compounds are typically optimized to nM or pM Kd values. Depending on the assay and tools available, Kd can be more challenging to interrogate and is less applicable in understanding a phenotypic effect, such as cellular cytotoxicity. When dealing with an inhibitor-target complex, the term inhibitory constant (Ki) maybe used in place of Kd. Ki generally requires even more intensive experimentation to determine, and involves measurement of compound inhibition across a series of inhibitor and substrate concentrations, in order to determine the intrinsic inhibitory activity for a particular enzyme target.
For inhibitors that compete with substrate for binding to the protein target, the Cheng-Prusoff equation can be used to convert IC50 values into Ki values, although the Michaelis constant (Km) should be a known variable.
Ki = IC50 / (1 + [S] / Km)
In general, it is often more feasible to determine IC50 values for all of the compounds in a library and ensure that the assay conditions remain constant. Notably, while lower Kd and IC50 values both indicate a more potent compound, due to the principles of thermodynamics, an experimentally determined IC50 cannot be lower than the Kd value. Therefore, it is not possible to keep reducing the concentration of substrate in an assay to artificially reduce the IC50 values observed.
3.3 Binding is Determined by Drug : Target Interactions
Ultimately, the capacity of a drug to engage with a target is determined by the thermodynamic binding energy. In a reversible reaction, the target enzyme (E) will bind an inhibitor (I) to give the complex E●I, where the equilibrium is governed by the constant Kd (or Ki in the presence of substrate). One of the main goals of medicinal chemistry and a significant portion of hit-to-lead investigations is to design molecules that will improve (decrease) the Kd. Understanding how to improve Kd, requires an understanding of the types of interactions that can occur between a target and its inhibitors. There are generally three types of binding interactions, i) electrostatic, ii) hydrogen bonding, and iii) “hydrophobic” interactions.
3.3.1 Electrostatic Interactions
Electrostatic interactions involve the interaction between two charged, or partially charged, species. These types of interactions represent the strongest type of non-covalent interactions. However, they are the most relevant in a hydrophobic environment. Within the environment of a polar solvent such as water, the charges may be partially of fully masked, as there is an increased opportunity for the ions to be solvated. Resultantly, the interactions between the charged residue and solvent (water) would need to be disrupted to facilitate inhibitor binding, which results in additional energetic binding penalties. In addition to location/environment, the intermolecular distance between the ions is also important as the strength is determined by Coulomb’s law of electrostatics. Therefore, lead molecules need to be optimized in order to position functional groups at ideal distances to target the appropriate residues on a protein. There are three main types of electrostatic interactions that are important in medicinal chemistry.
3.3.1.1 Salt-Bridges
Salt bridges typically form between positively and negatively charged species and play a large role in stabilizing compound binding. Salt bridges are the strongest type of electrostatic interaction (–20 to –40 kJ/mol). As with all electrostatic interactions, their strength relies on the distance between the two species. However, since these interactions are quite strong, they are the ‘least distance-dependent’ electrostatic interaction. Salt bridges generally dominate the initial long-range interaction between drugs and receptors.
Salt bridges largely depend on the dielectric constant of the media. The dielectric constant is a physical property that reports on the solvent polarity and varies substantially based on the environment. For example, the dielectric constant is ~80 for bulk phase water, 28 near the surface of a protein, and 4 at the interior of a protein (which is comparable to the highly non-polar organic solvents such as chloroform or dichloromethane). Salt bridges generally arise from specific amino acids – arginine, lysine, histidine (positively charged) or aspartic and glutamic acid (negatively charged) and will depend on the pKa of all amino acid side chains. For example, in the kinase inhibitor shown below (Figure 3.7), introducing an N,N-dimethylamino group in place of the isopropyl tail introduces a positively-charged nitrogen that is capable of forming a salt-bridge with the carboxylate group of Asp 831 on EGFR, leading to an ~1000-fold improvement in potency.
FIGURE 3.7 Replacement of the N,N-dimethylamino group enables a formation of a salt-bridge that increases potency.
3.3.1.2 Ion-Dipole Interactions
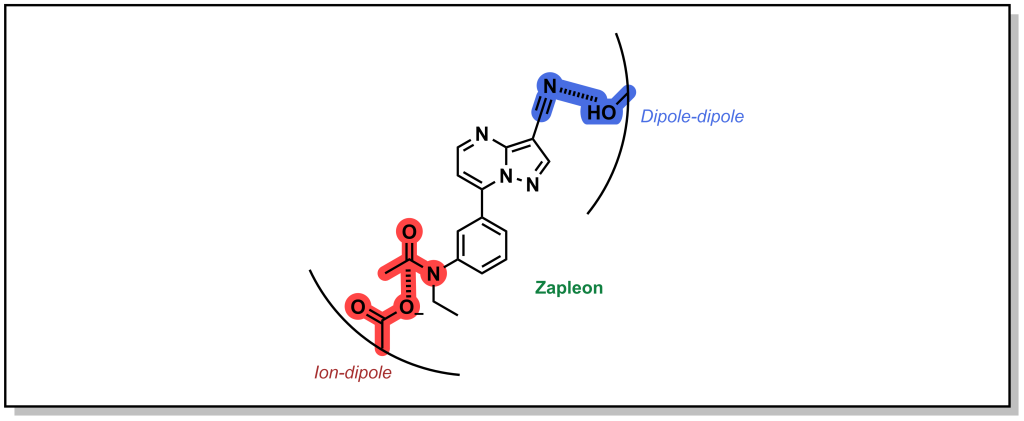
Ion-Dipole interactions result when one of the binding partners does not possess a full electrostatic charge. A dipole occurs when the electron density of a covalent bond is asymmetrically distributed as a result of the electronegativity difference between the two atoms (unequal sharing of electrons). This creates a directionality for a partial charge that can interact with a stronger ionic charge. The energy associated with ion-dipole interactions is –12 to –20 kJ/mol and decays with distance according to 1/r2 distance. Zapleon (Sonata) is a sedative used to treat insomnia that engages with GABA-A receptor via an anionic carboxylate sidechain which interacts with carbonyl centre of the drug in an ion-dipole interaction (Figure 3.8).
FIGURE 3.8 Structure of Zalepon with key ion-dipole and dipole-dipole interactions.
3.3.1.3 Dipole-Dipole Interactions
Dipole-dipole interactions are generally the weakest electrostatic interaction (–4 to –12 kJ/mol) but are the most common mode of interaction in drug-target binding and drive the affinity of many drugs for their target. Importantly, the magnitude of the dipoles is important, but the angle between the dipoles also affects the strength of the interaction. Certain angles are non-productive, and produce a zero value and depending on orientation. The energy of the interaction also decays with distance1(/r3). In the example above (Figure 3.8), the nitrile of Zalepon interacts with hydroxy-moiety of the side-chain in a dipole-dipole interaction.
3.3.2 Hydrogen Bonding
Hydrogen bonding involves sharing a hydrogen nucleus (proton) between an electron-rich heteroatom and an electronegative heteroatom. The electronic rich heteroatom (H-bond acceptor) must have a lone pair of electrons and is usually a nitrogen or oxygen atom. The H-bond donor is typically an electronegative heteroatom whose covalent bond to a hydrogen is highly polarized towards the electronegative atom (strong dipole). This also means, that generally speaking, carbon is not involved in H-bonding.
The free energy for H-bonding is usually within the rage of –6 to –30 kJ/mol and depends on the intermolecular distance and orientation of the H-bond. The shorter the H-bond, the more orbital overlap, and the stronger the interaction (typical H-bonds range from 1.5 to 2.2 Å). Similarly, the more linear the orientation, the greater the orbital overlap, with the maximum overlap occurring at 180º. Stronger H-bonds have a higher degree of symmetry which enables the H atom to be shared more equally.
Both oxygen and nitrogen are common hydrogen bond donors and acceptors, because they are electronegative atoms with lone pairs of electrons. Sulfur is a heteroatom, right under oxygen in the periodic table. However, sulfur is generally not an ideal hydrogen bond acceptor because its lone pair is in a 3s orbital which results in a large and diffuse electron cloud.
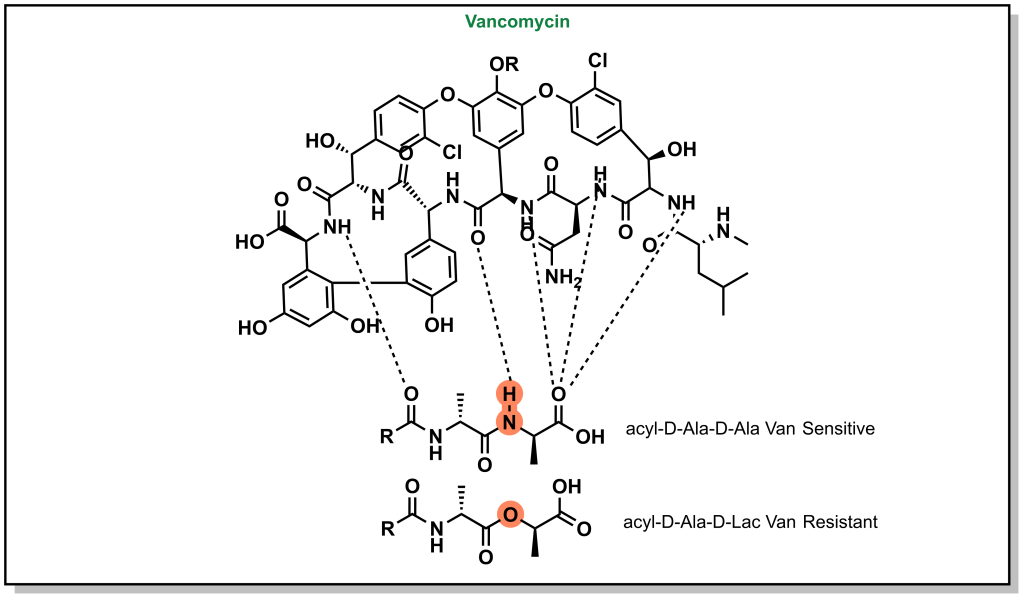
H-bonds can be critical to ensuring the proper inhibitor binding. Vancomycin is a non-ribosomal glycopeptide that acts by inhibiting cell wall synthesis in gram positive bacteria. It binds to a conserved sequence (acyl-D-Ala-D-Ala) at the end of the peptidoglycan chain and inhibits cross-linking. However, antibiotic resistance occurs when bacteria are capable of replacing the terminal D-Ala with D-Lac residue (lactate ester for an amide). This substitution disrupts a critical H-bond and leads to a 1000-fold reduction in affinity enabling bacterial survival (Figure 3.9).
FIGURE 3.9 Vancomycin forms critical hydrogen bonds with the peptidoglycan chain. Disruption of these H-bonds can result in resistance to vancomycin.
3.3.3 Steric Interactions – Van der Waals Interactions
Van der Waals interactions take place between non-polar molecules over very short distances (decay according to 1/r6). These molecules do not have a significant electrostatic attraction, but can result in transient dipoles, which can induce dipoles in other hydrophobic species. These are weak attractive forces (–2 kJ/mol), although the cumulative sum of these forces can be significant. Van der Waals interactions are common between the aliphatic groups of the molecule and also occur during aggregation of non-polar molecules in water or other polar solvents. There are also specific types of hydrophobic interactions that can occur in a subset of functional groups.
3.3.3.1 π-π Effects
Phenyl rings are found in 45% of all currently marketed drugs, partially because of the range interactions they can engage with, but also the known exit vectors of substituents, which allows for programmable optimization of the molecular structure. The electron density around an aromatic ring is delocalized and stabilized over the entire ring via resonance. The area above and below the plane of the ring is considered electron-rich and the movement of electrons through this system creates a quadrupole moment that can interact with cations (cation-π effect) and is shown to occur with positive charged residues (Lys/Arg) or aromatic residues (Phe/Tyr/Trp).
Alternatively, different types of π-π stacking can occur between closely positioned aromatic rings conformations. There are different types of π-π interactions, including sandwich, edge-to-face, and displaced π-π stacking. Although it is not a strict requirement, the highest attraction in π-π stacking occurs when the one of the π systems has an electron-donating substituent and another π system has an electron-withdrawing substituent (which creates pseudo-dipole interactions). Modern drug molecules nearly always harbour aromatic rings leading to critical π effects that are often stronger than conventional Van der Waals interactions. For example, crystallography analysis of an inhibitor library generated to target soluble epoxide hydrolase (sEH) reveals that introduction of an additional phenyl ring improves potency by 100-fold as a result of a new π – π interaction with His524 of the protein (Figure 3.10).
FIGURE 3.10 Introduction of an aromatic ring enables pi-pi stacking within the target soluble epoxide hydrolase.
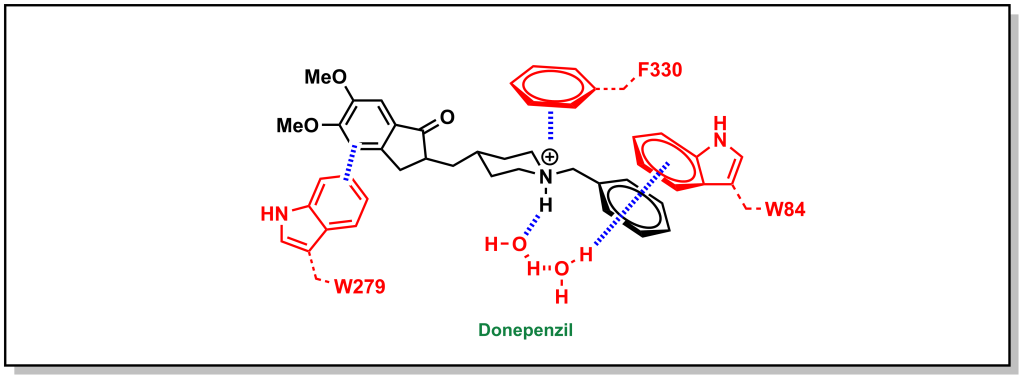
Similarly, the ACE inhibitor, donepenzil, participates in multiple aromatic interactions such as a cation-π interaction of the tertiary amine with Phe330, π-π stacking between the indole of Trp279 and the dimethoxybenzene, and π-π stacking of the Trp84 indole with the phenyl ring (Figure 3.11).
FIGURE 3.11 Donepenzil forms multiple aromatic interactions within the active site of Angiotensin-converting enzyme.
3.3.3.2 Halogen Bonding
Halogen bonding is a more exotic interaction that occurs with large, sigma-bonded halogen substituents. The sigma bond creates an asymmetry in the electron cloud focussed towards the equatorial axis of the halogen (with respect to the bond) and a partial positive charge directly opposite to the bond or a sigma hole. The sigma hole can act as an electrophilic centre and engage with electron-rich species in the trend I > Br > Cl. Importantly, fluorine is a special case and since it is quite small and electronegative, it does exhibit the properties of the sigma hole. Looking at phosphodiesterase 2 inhibitors, replacement of larger halogens demonstrated increasing potency via electrophilic interactions with the side-chain oxygen of Tyr827 (Figure 3.12).
FIGURE 3.12 Large halogens can introduce an electrophilic site on the molecule.
3.3.3.3 Magic Methyls
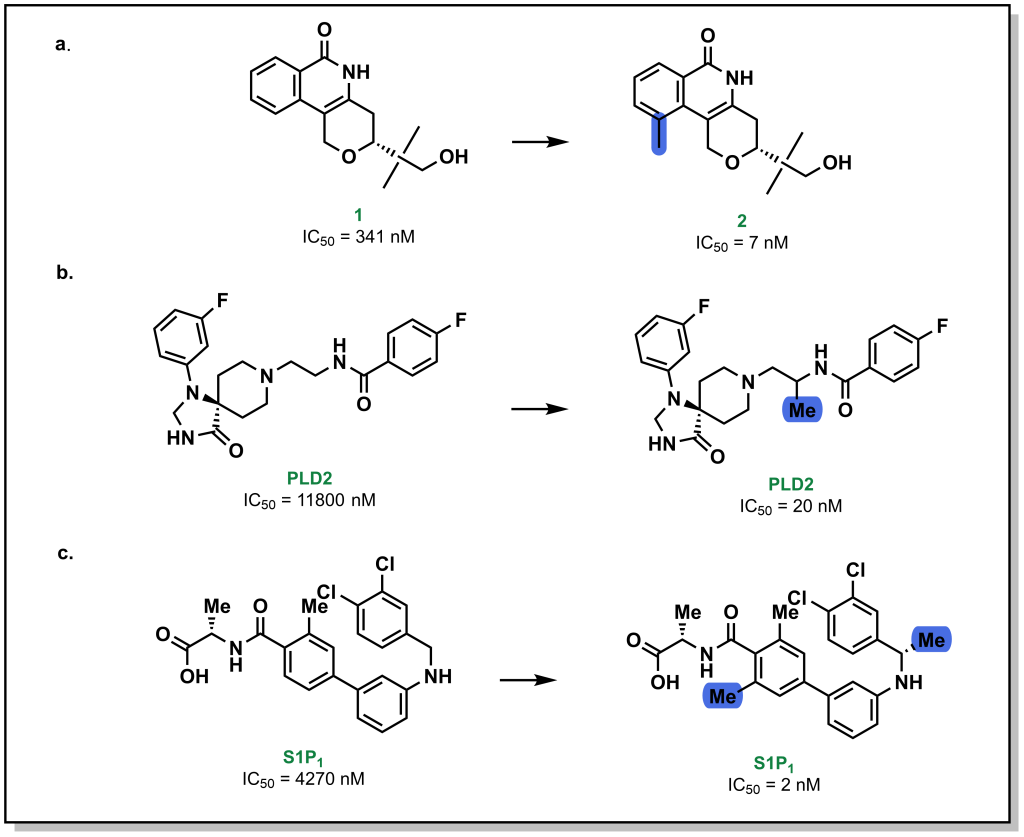
Methyl groups are small hydrophobic moieties that can drastically alter the affinity of a ligand for its target. For example, in investigating the SAR around tankyrase-2 (TNKS2) inhibitors, introduction of a single methyl group drastically increased the potency by ~50-fold (1 vs. 2; Figure 3.13a). Similar effects are observed for additional compound classes, such as PLD2 (Figure 3.13b) or S1P1 inhibitors (Figure 3.13c). The rationale for this substantial difference is often attributed to displacement of an unfavourably located water molecule in the binding site, which is both entropically favourable (water release) as well as enthalpically favourable (hydrophobic interactions of the methyl group). Additionally, methyl groups can also modify the dynamics of ligand to lock-in a favourable conformation. However, this is not a conserved effect and methyl group introduction can also lead to decreases in potency, particularly if the methyl group is not buried in a hydrophobic pocket upon protein-binding, or if an unfavourable conformation emerges.
FIGURE 3.13 Introduction of a methyl group at a specific site can yield remarkable increases in binding potency.
3.4 Energetics of Drug-Receptor Interactions
The interactions that are discussed above contribute to the Gibb’s free energy of binding (ΔG) for a ligand to its target. Thermodynamically, this can be explored as the enthalpy and entropy of the interactions based on the equation:
ΔG = ΔH – TΔS
From a superficial interpretation of physical chemistry, the enthalpy refers to the heat change upon binding and the entropy refers to the number of states that reaction can sample. For example, when a protein binds a ligand via dipole-dipole interactions, this is enthalpically favourable due to energy released by this interaction, but entropically unfavourable because there is one complex compared to the original two “free” species. However, monitoring energetics of these reactions can be more complex as there are multiple species in the system. For example, forming the dipole-dipole interactions may require de-solvation of the ligand or protein which would reduce the enthalpic gain of the reaction. Furthermore, binding of the ligand to the protein may release water molecules from the binding site of the protein that can lead to favourable entropic gains. These energetics are wholistically captured when determining the Kd of the interaction, and the relationship is characterized by the equation:
ΔGd = –RTlnKD
The equilibrium constant of dissociation (KD) is also related to the ratio of the rate constants for the on- and off- rates (koff/kon) for a drug which is critical for the concept of residence time. Although there are many tools to explore the energetics of compound-binding, one common theme is that if the free inhibitor is too flexible it will reduce Kd, since there will be a large entropic penalty upon protein binding. Therefore, a key part of optimizing compounds can involve reducing intrinsic conformational mobility through rigid systems (e.g. aromatic, sp2, or hetero-atoms)
3.5 Properties of Lead Compounds
Optimization of a lead compound requires strategic planning and consideration of all the potential drug-target interactions. When new compounds (hits or leads) are proposed and advanced the following questions are a useful starting point in gauging which properties of the molecule should be optimized:
What is the molecular weight of the compound?
Large compounds (>500 Da) are susceptible to low cellular permeability.
What hetero-atoms and functional groups are present?
Common heteroatoms include oxygen and nitrogen and include potential sites for protonation; certain functional groups can have metabolic liabilities or toxicities and can be replaced with bio-isosteres.
Are there any sp3 or stereocentres?
Stereocentres often complicate both the synthesis and the analysis of activity, but can potentially offer higher potency and more selectivity. Introduction of sp3 sites can be metabolic soft spots.
Are there any sites for protonation or ionizable sites?
Determining the pKa value for any sites of protonation can provide important information on the charged state of the compound in biological media.
What is the cLogP and tPSA (total polar surface area) of the compound?
The overall polarity and lipophilicity of the compound provides information on the predicted cell permeability.
How many rotatable bonds are on the compound?
Highly flexible molecules can be entropically penalized upon binding due to the reduced degrees of freedom and can thus reduce the binding affinity.
How many hydrogen bond donors (HBD) and acceptors (HBA) are present on the compound?
HBD and HDA can affect the polarity and reactivity of the molecule, as well as its metabolic stability, permeability, and efflux properties.
Answering these questions can provide a solid starting point for lead optimization and highlights physical properties that could potentially be downstream liabilities. A number of these properties were collated by Christopher Lipinski (who worked at Pfizer) in 1997 and developed the rule of five (Lipinski’s rule of five) which provides guidelines from analysis of orally administered drugs. Lipinski’s rules include:
Molecular mass of 500 Da (or less)
A cLogP of 5 (or less) [Oral drugs have a moderate lipophilicity of 2-5]
A maximum of 5 hydrogen bond donors (sum of OH and NH moieties)
A maximum of 10 hydrogen bond acceptors (sum of O and N atoms)
All of the rules involve parameters which are multiples of five, and provides the basis for the name. Lipinski’s rule of five was extremely influential in organizing compounds in medicinal chemistry and provided threshold goals for drug discovery programs. Although these rules provide crucial starting points, there are multiple drugs that defy these principles including antibiotics, antifungals, and glycosides (although some of these oral drugs have proteins that facilitate their transport across membranes). With the advent of new technologies such as bivalent degraders and protein-protein interaction inhibitors there has been contemporary emphasis on developing guidelines to go beyond the rule of 5 (bRo5).
3.6 Ligand Efficiency
Ligand efficiency (LE) is an important metric in evaluating different molecules in lead discovery. Ligand efficiency attempts to address how efficient a molecule is at engaging a target. For example, if there are two different candidates that both have a Kd of 10 nM, the smaller molecule (based on molecular weight) would be considered more “ligand efficient”: the atoms of the smaller molecule are engaging in more potent interactions, whereas the larger molecule requires more atoms to achieve the same potency. This suggests that some of the additional atoms could be removed if they are not contributing to the potency and offers a starting point for medicinal chemistry iterations. The formula for calculating ligand efficiency is shown below:
LE = ΔGd / #HA
Remember that ΔGd is related to the Kd which directly reports on target-ligand binding. The term ‘heavy atom’ (HA) in this context refers to any atom that is larger than hydrogen (C, N, O, S, etc.). In general, higher values for ligand efficiency are associated with a more drug-like compound, although this is not without controversy. For example, consider the two drugs captopril (an ACE inhibitor used for the management of hypertension) and rosuvastatin (also referred to as the cholesterol lowering agent, Crestor, an inhibitor of HMG-CoA reductase). Captopril has 14 heavy atoms, whereas atorvastatin has 33 heavy atoms. The Kd of captopril to ACE is 8.5 nM and the Kd of rosuvastatin to HMG-CoA reductase is 2 nM. The ligand efficiencies for captopril and rosuvastatin are therefore 3.4 kJ/mol/HA and 1.1 kJ/mol/HA respectively. Although captopril has a higher ligand efficiency, both drugs are widely used and this provides an example of the debate in optimizing drugs based solely on ligand efficiency.
Drug Absorption and Distribution
4
“Science is magic that works.”
– Kurt Vonnegut
4.1 Introduction to Pharmacokinetics and Pharmacodynamics
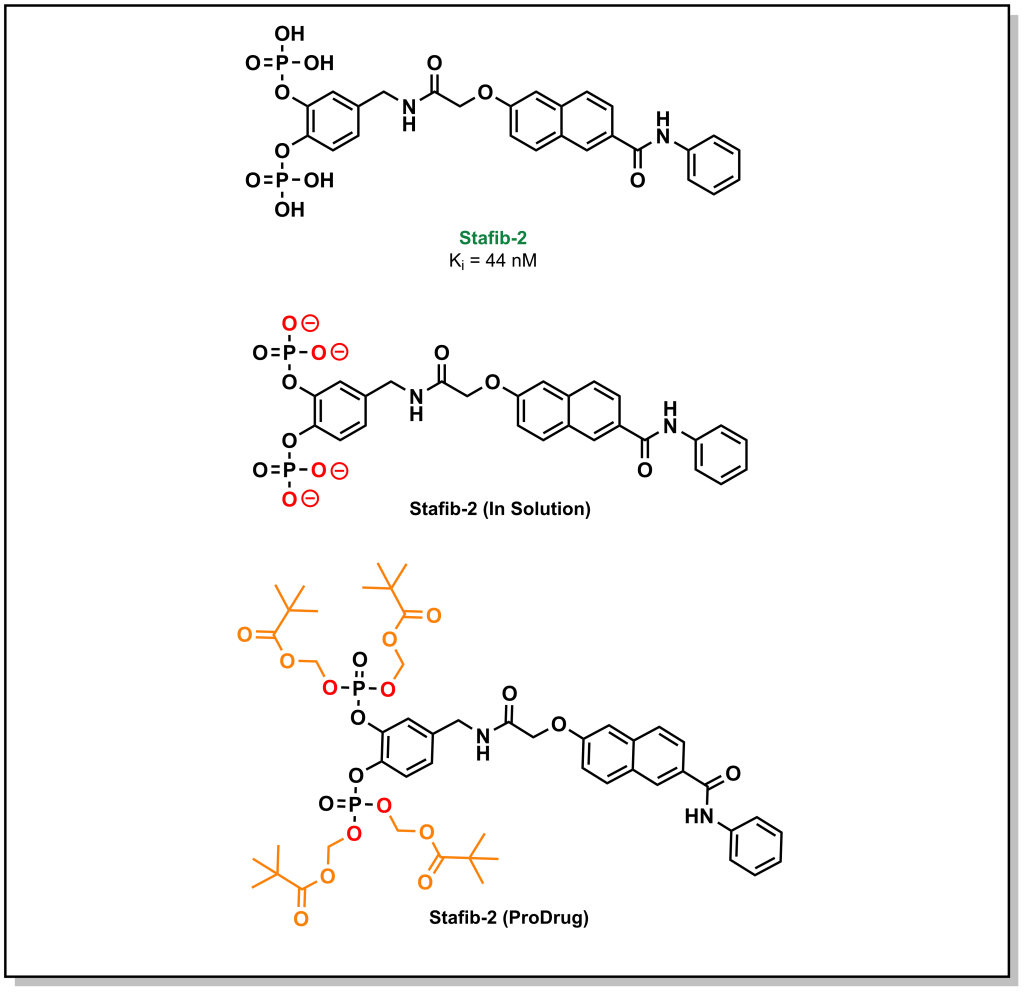
A large emphasis of the early stages in drug discovery is placed on obtaining a lead molecule that can elicit a potent effect on a target of interest. Once a lead is generated, the next stage involves exploring the pharmacological utility of the molecule. For instance, a lead may exhibit high potency and selective target engagement in vitro but is not capable of exerting a biological effect in cellulo or in vivo. The molecule, staffib-2, provides a representative example of this conundrum. Stafib-2 was designed to target the SH2 domain of STAT5B, a transcription factor upregulated in multiple cancers. The origins of the molecule emerged from natural products and catechol bisphosphate derivatives, which can mimic phospho-tyrosine moieties (SH2 binding scaffolds). The structure is chemically optimized to exploit a π-π stacking interaction of the catechol with a tryptophan in the SH2 domain, along with hydrophobic interactions of the transposed benzamide with a nearby phenylalanine (Figure 4.1). The resulting molecule demonstrated potent inhibitory activity (Ki = 44 nM) with >50-fold selectivity over the closely related STAT5A isoform. Based on these data, this is an excellent hit molecule, but it demonstrates extremely poor activity in cellular assays. This lack of activity arises from the pKa values of the phosphate groups (1.5 and 6.3), which are indicative of the ionization state of the molecule at physiological pH. The di-anionic charge on each phosphate group prevents the molecule from effectively crossing the phospho-lipid bilayer, and these properties had to be masked in the form of a pro-drug in order to obtain phenotypic efficacy.
Figure 4.1 Masking anionic charges on stafib-2.
Therefore, it is not always sufficient to identify a molecule with excellent potency and selectivity. The effects that physiological processes of the body can have on the drug, as well as the effects that the drug can have on the body need to be considered. These effects are called pharmacokinetics (PK) and pharmacodynamics (PD), respectively. Understanding and optimizing both the PK and PD properties of a molecule forms a large component of medicinal chemistry and the drug discovery pipeline.
4.2 DMPK & ADME
Drug Metabolism and Pharmacokinetics (DMPK) refers to the study of how the body will engage and interact with a drug. PK is extremely important in drug discovery, as a molecule that enters the body will be treated as a foreign entity. Hence, it will need to survive the multiple defense mechanisms that the human body has evolved to deal with xenobiotics, in order to reach its specific target and achieve therapeutic effects. Pharmacokinetics is often sub-divided into different processes which include drug Absorption, Distribution, Metabolism, and Elimination, and are collectively referred to by the acronym ADME. Although ADME is often presented and treated as distinct phases of the overall DMPK process, these phases are all interrelated. The focus for medicinal chemistry will be towards absorption and metabolism whereas distribution and excretion are largely biology/physiology-based topics.
4.3 Absorption In Cellulo
Initial exposure to a drug and its uptake into the body is called absorption. There are different considerations for drug absorption that depend on the complexity of the biological and molecular system under evaluation.
4.3.1 Cellular Membranes
For an in cellulo experiment, the molecule is administered directly to the local environment of the cell. If the target exists on the outer layer of the cell surface (e.g. a GPCR), the molecule does not need to transverse the cell membrane and can effectively bypass a number of permeability challenges. However, if the target exists within the cell, a drug will need to cross the membrane to reach its intracellular target. Recall that the structure of a cell membrane is formed by a phospholipid bilayer intercalated with proteins. As such, the negatively charged phospho-lipid headgroups are facing both the extracellular and intracellular matrix and create a highly non-polar “sandwich” that segregates the cell from exterior contents. The biochemical implications of this bilayer structure impose unique property requirements for drugs/compounds traversing the membrane.
4.3.2 Transport – Passive Diffusion
Passive diffusion refers to the spontaneous movement of a substance from a region of high concentration to a region of low concentration. For a drug that is administered, this means that (at least initially) there is a high concentration outside the cell and a low concentration inside the cell. Based on concentration gradients alone, a drug would therefore be energetically favoured to enter the cell. However, polar or charged molecules cannot readily partition into the hydrophobic lipids of the bilayer and diffuse at a substantially lower rate. This can be quantitively described by Fick’s law of diffusion.
As a general trend, neutral compounds will have a higher rate of passive diffusion, followed by bases, zwitterions, and acids. Passive diffusion is the predominant mechanism of transport for lipophilic compounds. Although bases carry an ionic charge, they are not as energetically disfavoured for passive diffusion since the positive charge of the molecule can associate with the negatively charged phospho-lipid headgroups to initiate the diffusion process. Similarly, zwitterions (positively and negatively charged species) can also associate in this manner and have a net neutral charge. Acids (negatively charged species) are especially challenging to transverse the membrane via passive diffusion due to repulsive electrostatic interactions with the phospholipid headgroup. However, this impermeability of anionic species to the lipid bilayer can be an effective tool in drug discovery. Consider the example of Stafib-2 above. The negatively charged phosphate groups prevent the uptake of the compounds into the cell interior. These charges can be masked by generating phosphate esters. This creates a neutral species that is now capable of crossing the membrane via passive diffusion. However, Stafib-2 needs to be negatively charged to engage with the positively charged sub-pocket of the STAT5B SH2 domain. Esterases (which are located predominantly within the intracellular matrix) can hydrolyze the compound into its active anionic form once it enters the cell. This negatively charged species can now engage with the target. Furthermore, the negatively charged species becomes trapped within the intracellular compartment and cannot passively diffuse out of the cell. Hydrolyzing the ester also reduces the concentration of drug-ester inside the cell continuing to pull the equilibrium towards compound influx. This strategy of generating an inactive species that is later converted to an active form, is a called a pro-drug approach.
4.3.3 Transport – Facilitated Diffusion
As depicted above, not all drugs (or organic compounds) are amenable to passive diffusion. Cells have developed multiple strategies for the transport of compounds that cannot be readily absorbed because they are too large or too polar. This includes a number of transporter proteins that can be hijacked by drugs for cellular import. These specialized transport mechanisms can generally be stratified into two streams, facilitated diffusion and active transport. Notably, both of these processes rely on a transporter, and therefore the protein can be saturated which can limit transport at high concentrations.
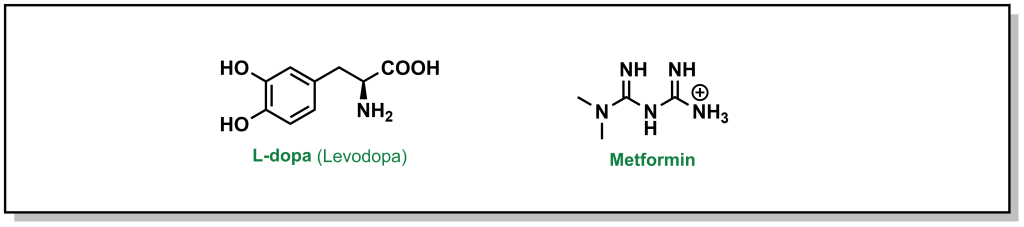
Facilitated diffusion is a passive process, where a molecule will move down a concentration gradient, but it requires a protein to mediate transport. As such, this is a selective process, as only specific chemotypes are permitted passage. Different drugs can leverage the use of known transporters. For example, L-dopa (Levodopa) is a drug used for the treatment of Parkinson’s disease that is too polar to passively diffuse across the blood brain barrier membrane (Figure 4.2). The amino acid structure of the drug can exploit the LAT1 transporter (L-type amino acid transporter) to enter the cell. However, utilizing natural transporters can also lead to challenges, as dietary consumption of protein sources (ie. amino acids), can compete with L-dopa and saturate the transporter, indicating patients have to monitor the timing of L-dopa dosing with their meals. Another example involves the most common blood glucose lowering agent for Type II diabetes, metformin (Figure 4.2). Metformin is a positively charged biguanide that cannot passively cross the membrane. Metformin enters cells via the Organic Cation Transporter-1 (OCT-1) which enables transport of positively charged species.
FIGURE 4.2 L-dopa and metformin
4.3.4 Transport – Active Transport
In contrast to passive or facilitated diffusion, active transport requires energy input to transport a molecule against its concentration gradient. The energy is provided in the form of ATP hydrolysis or an electrochemical/concentration gradient. In drug discovery, active transport is most relevant in terms of the efflux of xenobiotics (drugs) out of the cell. The Multi-Drug Resistance (MDR) proteins are a family notable for their ability to mediate the effluxing of drugs from cells, preventing their cellular accumulation, and augmenting disease cell survival.
Multi-drug resistance can be an especially challenging aspect in drug discovery and represents a general mechanism where the lifetime of a drug inside a cell is not extensive enough to carry out its therapeutic effects. It is important to note that this is different from specific types of drug resistance that can result from mutation of a protein to prevent action of the drug on its target (eg. the gatekeeper mutation in EGFRT790M discussed in Section 1, blocks the action of the kinase inhibitor, afatinib).
Although it is non-obvious whether a molecule is a substrate for an efflux-transporter, there are considerations to help limit drug-interactions with transporters. In general, efflux pumps effectively transport molecules with the following properties out of the cell:
Lipophilicity
Planar Structures
Molecular Weight < 800 Da
Weakly cationic
Unfortunately, these are also heavily desired properties for small molecule inhibitors. There are five protein families from the drug/metabolite transporter (DMT) superfamily.
ATP-binding cassette (ABC) Transporters: This is one of the largest and most ancient protein superfamilies. The architecture of a conventional ABC transporter includes 2 transmembrane domains and 2 nucleotide binding domains that bind and hydrolyze ATP to facilitate conformational changes and pass substrates through the membrane. ABC transporters have diverse functions, and a subset of these proteins have drug efflux capacity. Multidrug resistance protein (ABCC1 or MRP1), and breast cancer resistance protein (ABCG2 or BCRP) confer resistance to several drugs. However, the most notable example is ABCB1, also called P-glycoprotein (P-gp) or MDR1, which poses a large obstacle in overcoming drug resistance for a broad array of structurally-diverse compounds (see below). P-gp is the most relevant drug efflux protein and specific assays evaluating P-gp activity are often incorporated into drug discovery pipelines.
Major Facilitator Superfamily (MFS) or Uniporter-symporter-antiporter family: Similar to ABC transporters this is the second largest family of membrane transporters and is found in bacteria, archaea, and eukarya. Together with ABC transporters, these groups encompass more than half of all known membrane transporters. Unlike ABC transporters, these proteins rely on electrochemical gradients to facilitate transport and are usually single-polypeptides.
Multiple Antimicrobial Extrusion Family (MATE) transporters: This is a class of secondary active transporters that are largely involved in transport of cationic compounds and are present in all domains of life. MATE transporters are often attributed to multi-drug resistant hospital infections due to their expression in Staphylococcus aureus. In humans, these transporters are mostly present at the brush-border of the kidneys. Similar to MFS transporters, typical MATE transporters contain 12 transmembrane helices.
Resistance-nodulation-division (RND) Transporters: These transporters are found in (Gram-negative) bacteria as well as archaea and are particularly important in microbial xenobiotic defense/efflux. These pumps are asymmetric trimers that derive energy from proton gradients.
Small Multidrug resistance (SMR Transporters): These are bacterial transporters comprised of four alpha helices that also derive energy from electrochemical proton gradients. These transporters focus on the movement of lipophilic compounds and quaternary ammonium compounds.
4.3.4.1 P-glycoprotein (P-gp)
P-glycoprotein is the most clinically relevant drug transporter due to its broad specificity and presence in the key tissues for ADME including the gastrointestinal tract, blood-brain-barrier, kidneys, and liver. P-gp acts in a unidirectional manner to extrude substrates outside of the cell, which can effectively topple a lead candidate in drug discovery pipelines. There are extensive published lists of drugs that are known to be actively effluxed by P-gp and 25% of all oral lipophilic drugs are known P-gp substrates.
P-glycoprotein affects the rate and concentration of drugs diffusing across the basolateral membrane of the intestine. For example, in an enterocyte (cells along the small intestinal lining), P-gp is expressed at the brush-border (site of absorption). This facilitates the removal of xenobiotics almost immediately and prevents their absorption. Another key site of P-gp expression is at the blood-brain-barrier (BBB), which represents a number of tightly packed cells that surround the central nervous system (CNS). These endothelial cells of the BBB contain P-gp that effluxes compounds and presents a major challenge in drugs accessing the CNS tissues. In drug-discovery pipelines, P-gp efflux is often evaluated through an MDCK1-MDR1 assay which involves Madin Darby Canine Kidney (MDCK1) cells transfected with the human MDR1 (P-gp) gene grown in a monolayer. An efflux ratio of < 2 indicates the compound is not a substrate for P-gp.
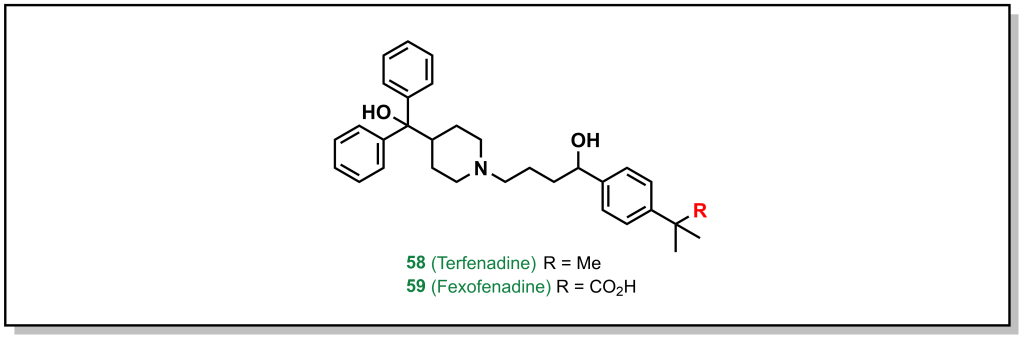
Amgen’s medicinal chemistry analysis of 4176 drugs demonstrated that 52% of P-gp substrates showed tPSA > 90 Å2 and 57% of P-gp substrates showed > 2 HBDs. Additionally, highly lipophilic drugs with LogP > 7 were P-gp substrates. These trends provide potential approaches to evade P-gp efflux, which are focussed on reducing HBD character as well as acidic/basic groups. Substrates that contain nitrogen atoms found in amines, amides, sulfonamides, and heterocycles, as well as oxygen atoms in hydroxy and carboxylic acids are also alerts for P-gp efflux. Although it is not a strict guarantee that molecules with these functional groups will be effluxed, if it is possible to remove the offending functional group without significant loss of activity, this is the best option. For example, removal of the carboxylic acid group from fexofenadine reduces P-gp efflux in terfenadine (anti-histamines for allergy medication; Efflux Ratio = 2.9 [Terfenadine] vs 6.8 [Fexofenadine], Figure 4.3). However, this is not always possible, in which case altering the hydrogen bond donor needs to be examined, such as: i) The proton can be directly removed (such as via alkylation) or ii) it can be masked. There are an array of different strategies to accomplish this for each functional group.
FIGURE 4.3 Removal of the acid reduces P-gp efflux.
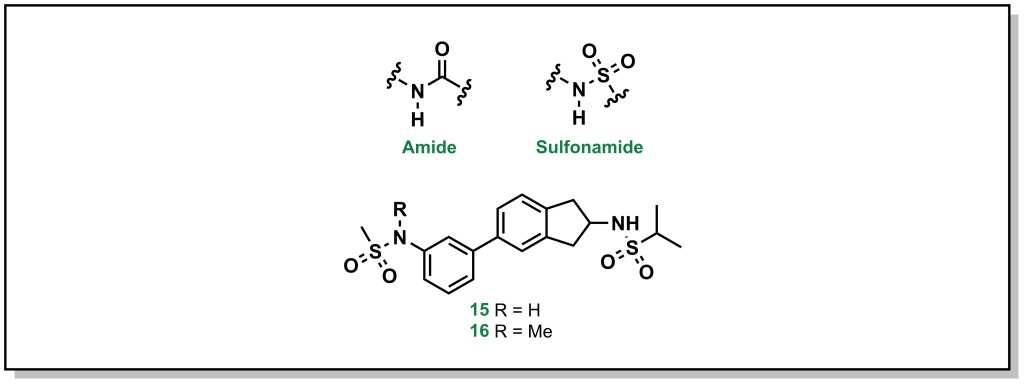
4.3.4.1.1 Amides and Sulfonamides
Amide and sulfonamide groups have a free N-H that is capable of functioning as a hydrogen bond donor. In specific substrates, it is straightforward to simply remove the proton by methylation, as with the AMPA receptor inhibitors developed by GSK (Figure 4.4). The parent compound (1) demonstrated a P-gp efflux ratio of 5.8, and upon methylation of one of the sulfonamides (2) the ratio dropped to 3.2 (remember that < 2 indicates the molecule has limited P-gp efflux).
FIGURE 4.4 AMPA receptor inhibitor.
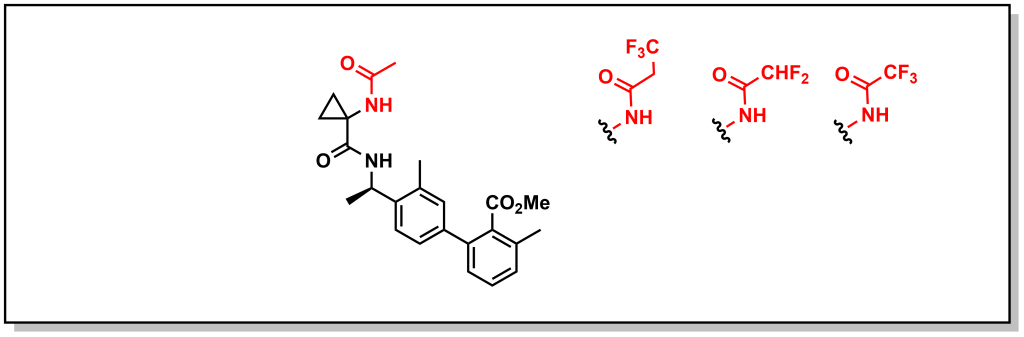
However, removal of the proton is not always tolerated and in such cases reduction of HBD character is beneficial. This can involve introducing an electron withdrawing group to reduce the HBD capacity of an amide/sulfonamide by removing electron density from the N-atom and modulating the pKa. For example, another series of bradykinin B1 receptor antagonists developed by Merck was shown to be substrates for P-gp. Careful manipulation of the alpha-amide substituents by appending fluorine atoms inductively reduced electron density from the amide and reduced P-gp efflux (Figure 4.5).
FIGURE 4.5 Bradykinin B1 receptor antagonists.
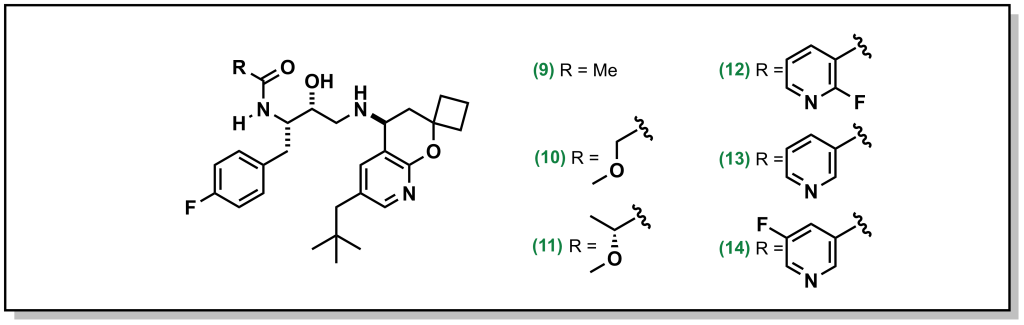
Other creative strategies involve masking the H-bond donor such as locking a pre-formed H-bond. For example, BACE (β-Amyloid cleaving enzyme-1) inhibitors are a target in Alzheimer’s disease that were pursued by Amgen. However, the amide proton was shown to facilitate P-gp efflux. Installing R-groups with a hetero-atom facilitating an intramolecular H-bond with the amide proton, masked the availability of the HBD thereby reducing P-gp efflux (Figure 4.6). For example, the ether linkage demonstrated correct positioning for intramolecular H-bond formation. Interestingly, the fluorinated pyridine showed reduced P-gp activity, which was not simply case of increased bulk around the H-bond (the non-fluorinated pyridine still demonstrated P-gp efflux). Instead, the fluorine atom of the pyridine is positioned to participate in an H-bond. This is a creative strategy since H-F bonding is weaker than H-O or H-N, but was structurally confirmed to participate in an H-bond and is successful at blocking P-gp activity.
FIGURE 4.6 Altering the acidity of BACE inhibitors via the R group.
4.3.4.1.2 Amines and Heterocycles
Basic amines and heterocycles can also impart P-gp efflux onto drugs, and reducing the pKa of the amine can improve activity (similar to above). For example, orexin antagonists developed by Merck for narcolepsy treatment contain imidazole analogues. Although the parent compound demonstrated an efflux ratio of 13, direct methylation of the imidazole removes the proton and dropped the efflux ratio to 2 (Figure 4.7).
FIGURE 4.7 Removing acidic protons or reducing their pKa can improve the metabolic stability of amine containing compounds.
In cases where removing or blocking the heteroatom is not possible, reducing the pKa is also feasible. For example, the PXR activator was shown to have low metabolic stability which was improved by installing of the piperidine ring. However, the amine resulted in P-gp efflux, and switching to a morpholine ring inductively reduced the pKa of the nitrogen and subsequently the P-gp efflux (Figure 4.7).
4.3.4.1.3 Alcohols and Carboxylic Acids
Alcohol and carboxylic acids follow the same principles outlined above, and the most common strategy is removing the proton through alkylation. For example, tolerance to morphine is thought be partially imparted by increased expression of P-gp (Figure 4.8). Likewise, carboxylic acids can be converted into other substrates functionalities such as esters, carboxamides, or dimethylformamides, which can also facilitate in masking the anionic charge and improving cellular permeability.
FIGURE 4.8 Modification of morphine via alkylation.
4.4 Absorption In Vivo
The above absorption parameters predominantly only consider cellular permeability. Within more complex living systems, additional factors need to be considered. For an in vivo experiment, the route of administration affects the rate of absorption and effective concentration as the drug is required to survive the journey through different organs and tissues of the body to find its target. Some of the different routes of administration are shown in Table 4.1. The main focus will be on IV, PO, and IP, although the FDA has over 100 approved administration routes.
4.4.1 Intravenous Administration
In intravenous (IV) administration, the drug is administered directly into the veins via a needle. This provides a rapid onset of action because the drug directly enters the blood stream without passing through any organs and there is no barrier to absorption. Drugs may be administered as a bolus (single dose) or infusion (continuous drip). Once the drug enters systemic blood circulation it is distributed throughout the body. A critical point of IV administration is that it avoids first-pass metabolism. This refers to metabolism of the drug at tissues in the body before it reaches its site of action. For example, remdesivir is an antiviral drug that is administered by IV injection. If it was administered orally, it would be subject to metabolism in the liver where it would be chemically altered before it had a chance to reach the intended site of action. Although IV administration allows for rapid onset, precise control of dosing, and avoids initial metabolism, it is not the preferred route for drug delivery because of the inconvenience and training required for administration of the drug.
4.4.2 Oral Administration
Oral formulations (pills, gels, liquids) are the preferred route, partly because of the ease of administration and patient compliance. However, the drug is taken by the mouth which means it passes through the stomach, small intestine (allowing absorption into the blood), liver, right atrium/ventricle of the heart, and then into full circulation. As such, there is generally a delay in onset as action, as the drug needs to pass through several organs before it enters systemic circulation. Furthermore, the drug must survive the acids/enzymes of the stomach, achieve absorption via the small intestine, survive metabolism in the small intestine and metabolism in the liver. Unsurprisingly, due to these hurdles in absorption and metabolism, not all of the drug that is administered orally will enter systemic circulation so oral doses are usually higher than IV doses. The amount of drug that reaches the bloodstream can be compared the amount of drug that enters the bloodstream if the patient was given the drug via IV administration, and this is represented quantitatively as %F which is called the bioavailability. A low %F indicates that the drug has poor bioavailability and is either poorly absorbed, rapidly metabolized or both, but does not always preclude a drug from therapeutic usefulness. The bioavailability of aspirin is ~50% and morphine as a bioavailability of ~30% as these drugs are metabolized in the liver. Warfarin is blood thinning agent whose %F is >99% which is exceptionally unusual for a drug.
4.4.3 Intraperitoneal Administration
Intraperitoneal administration is important in preclinical models and trying to isolate challenges in bioavailability. After injection, the drug is ultimately absorbed in the superior mesenteric vein (meeting up with other orally administered nutrients/drugs/substances that come from the small intestine capillaries via the jugular vein), which converges with the hepatic portal vein and enters the liver. This indicates that drugs administered via IP still undergo first pass liver metabolism, although absorption/degradation by the stomach and small intestine does not occur.
4.4.4 Pharmacokinetic Parameters
Quantitative pharmacokinetic profiling of a drug is usually done by analysis of the blood using mass spectrometry. Following administration of a specific drug dose (via any route), the blood is sampled at different time points. The sample is processed and analyzed by mass spectrometry to determine the concentration of drug in the blood at that time point. Plotting the concentration of compound over time allows for a number of important pharmacokinetic parameters to be determined:
Cmax: This is the maximum concentration of drug in the blood that is achieved throughout the entire study. This provides an estimate of how well the drug is absorbed.
Tmax: This is the time at which Cmax occurs. This provides an understanding of how long absorption takes.
t½: This is the half-life which reports on the how fast the drug is eliminated from the blood.
AUC: This is the area-under-the-curve which is the integral of the concentration of drug vs. time. This provides an estimate of the total exposure of the body to the drug and helps to put Cmax and t½ into perspective. Two potential drugs could have the same Cmax but if one candidate has a double the AUC value, that means this body has twice as much exposure to the drug.
Optimizing these parameters can vary between each drug. For example, using a drug that has a short-half life can be challenging, since it might be completed eliminated. However, if the drug has a very long half-life there can be toxicity side effects. Therefore, the pharmacodynamic effects also need to be considered when evaluating these parameters.
The PK profile for an IV administered drug usually demonstrates a Cmax at the initial timepoint since the drug is injected directly into the blood. As time passes, the drug distributes across different tissues and the blood concentration is reduced. For an orally administered drug, the drug has to pass through several organs before entering the blood stream and Cmax usually occurs later in the PK profile.
4.5 Distribution In Vivo
Following absorption, the drug enters systemic circulation and can access different parts of the body, including the site of the target. In the absence of any confounding variables (such as interactions with tissues or proteins), the drug would continue to circulate in the bloodstream and end up passing through the liver and kidneys with each cycle, and be successively metabolized or cleared until the drug is fully eliminated. However, a number of additional biological processes occur during distribution.
4.5.1 Whole Blood and Plasma Protein Binding
The first aspect to consider in distribution is the composition of fluid that harbours the drug, i.e. whole blood. Whole blood is a mixture of blood cells (red blood cells, white blood cells, and platelets that make up ~45% of the volume of blood) and plasma (~55% of whole blood volume). Plasma is fluid that contains water, electrolytes, and proteins (with the most common plasma protein as albumin at ~45 mg/mL). The high concentration albumin makes it a key target for engaging with drugs in circulation, and its common to have both a drug fraction-bound and drug fraction-unbound. The free-drug hypothesis states that the concentration of free-drug in the plasma (as opposed to total drug concentration) determines the therapeutic activity. Therefore, only the unbound fraction or free drug is available to carry out therapeutic effects. From this, it can be inferred that a drug that is administered and is highly bound to plasma proteins may not be as effective. For example, the blood thinning agent drug warfarin has an observed bound-fraction of 97% (this is actually favourable for warfarin since as a blood thinner, the predominant site of action for warfarin is the blood). Albumin is largely positively charged, and acidic or neutral drugs display a higher propensity for binding. Basic drugs can exhibit binding to the negatively charged alpha-1 acid glycoprotein.
Plasma protein binding can also lead to different effects that can further complicate drug distribution. For example, once the drug binds to plasma proteins, it act as a reservoir for drug: as drug reaches its target site and diffuses into the appropriate tissues, the concentration of free drug in the bloodstream will be reduced, and Le Chatelier’s principle indicates that bound-drug will be released to maintain a dynamic equilibrium. Alternatively, if a patient is administered two drugs that exhibit plasma-protein binding, these drugs may compete for the same binding sites on the plasma proteins. As such, the drugs may exhibit lower apparent plasma protein binding and this can lead to a higher effective dose (ie. unbound drug) realized in the patient, than if the two drugs were administered separately.
4.5.2 Volume of Distribution
In addition to distribution within whole blood, the drug will also partition into different tissues. This is often described through ‘compartment analysis’. The body can be thought of as different compartments, where a compartment refers to spaces/tissues with similar properties (and not necessarily separate organs). For example, blood can be assigned one compartment and all non-CNS tissues can be classified as a second compartment. Additional compartments can be incorporated depending on the extent of the distribution analysis.
The distribution of a drug is given by the apparent volume of distribution (Vd). A larger volume of distribution indicates the drug does not remain in the blood and enters other tissues or compartments. Conversely, a lower volume of distribution indicates the compound remains the blood. Therefore, a higher dose would be required for a drug with a high Vd to obtain the same plasma concentration as a drug with low Vd. For drug administration, Vd is an important parameter for to determine the dose to be applied to a patient.
Importantly, the Vd is not a real physical volume but provides an understanding of drug accumulation in the body. The average patient weight is 70 kg and the total volume of water in the body (~70%) is 49 L. Furthermore, there is approximately 5 L of blood per person, and since ~55% of the total blood is plasma, there are ~2.75 L of plasma in a patient. Based on these numbers, a drug with Vd > 40 L is thought to be distributed throughout all of the tissues in the body. Some drugs such as chloroquine (Vd = 15,000 L) or atorvastatin (Vd = 420 L) are well over the actual physical volume of the body. These high Vd values indicate that the compound highly concentrates in tissues in the body and does not remain within the blood. Chloroquine is an antimalarial drug that is highly lipophilic which helps explain its high Vd. Atorvastatin is also lipophilic and is known to accumulate in muscles which is partially responsible for some of its observed side effects. By contrast, warfarin and ibuprofen have low Vd values of 10 L and 10.5 L respectively, indicating they remain the blood. As the volume of distribution increases, the half-life of the drug also becomes longer. This is because the drug is distributed across more tissues which lead to additional time required for undergo renal or hepatic clearance.
Drug Metabolism
5
“I can’t change the direction of the wind, but I can adjust my sails to always reach my destination.”
– Jimmy Dean
5.1 Biological Pathways for Drug Metabolism
Following administration, drugs are absorbed and distributed into different tissues. Over time, these molecules are eliminated (irreversibly removed) from the body which can occur in two ways: either directly unchanged (excretion/elimination) or changed (metabolized). Drug elimination predominantly occurs through the kidneys (especially for hydrophilic species) and exits via urine, although a subset of drugs (predominantly non-polar substances) are eliminated in the bile (via action of the liver) and excreted in the feces.
The liver is the principal site for drug processing and metabolism and has (without exaggeration) hundreds of characterized functions including roles in digestion, detoxification, immunity, hormone release, ammonia processing, glycogen production, and heavy metal storage. The liver also produces bile, which is a mixture comprised of sterol derivatives (bile salts), cholesterol, phospholipids, and electrolytes, that is stored in the gall bladder and released in the small intestine to facilitate digestion of fats. This hydrophobic character of bile also becomes important in the metabolism of highly non-polar drugs.
The liver holds approximately 10-15% of the total blood supply at any moment, and all blood that departs the stomach or intestines passes through the liver enabling absorption of nutrients (or metabolism of xenobiotics in the first-pass effect). The liver is comprised of functional units termed lobules, which are typically hexagonal structures characterized by a hepatic portal vein, artery, and bile duct at each hexagonal vertex. The portal veins drain into a central vein at the focal point of the hexagonal lobule, that carries blood away from the liver into systemic circulation. These veins (as well as the ducts and arteries) are lined by parenchymal liver cells which are called hepatocytes. Hepatocytes comprise approximately 60-70% of all the cells in the liver and have an asymmetric cellular structure with the periportal side (Zone 1) facing oxygenated blood/nutrients. The cellular face furthest away from the portal triad is called Zone 3 and plays the largest role in detoxification and biotransformation of drugs. The space sandwiched between these zones (pericentral region) is intuitively referred to as Zone 2 of the hepatocyte.
Importantly, the flow of blood through the veins and the flow of bile through the ducts occur in opposite directions to each other. This is consistent with the generation of both substances as bile is produced in the liver, whereas blood enters the liver for perfusion. As mentioned, blood drains from the branches of the hepatic veins into the centre vein of the lobule.
5.2 Phase I Metabolism
Drug metabolism refers to the chemical modification of a drug, and the overall goal is to generate a more polar or water-soluble derivative that is amenable to elimination. Drug metabolism is generally classified as either Phase I or Phase II, although these do not need to be successive steps, and some drugs will only be processed through one of these two phases of metabolism. Both phases of metabolism occur in the hepatocytes of the liver.
Phase I metabolism is characterized by functionalization of the drug via one or more of the reactions:
Oxidation (electron removal, dehydrogenation, or oxygenation)
Reduction (electron addition, hydrogenation, or removal of oxygen)
Hydration/dehydration (hydrolysis, and addition or removal of water)
These transformations are carried out in the endoplasmic reticulum of hepatocytes and the most prominent Phase I reactions are oxidations. In an ex vivo setting, hepatocyte cells can be disrupted and processed by equilibrium density centrifugation to isolate vesicle-like pieces of the endoplasmic reticulum which are called microsomes. Although microsomes do not exist naturally, they provide a convenient tool to analyze Phase I metabolism. Over 75% of Phase I metabolism is performed by a class of enzymes called Cytochrome P450 (CYPs or P450) enzymes. These enzymes are heme-coupled monooxygenases and they bind oxygen (as well as carbon monoxide which leads to a wavelength absorption at 450 nm and serves as their namesake).
5.2.1 Cytochrome P450 Enzymes
There are at least 57 CYP enzymes encoded within in the human genome, with CYP3A4 and CYP2D6 having the highest abundance and contributing to >50% of all CYP-related metabolism. The nomenclature for these enzymes is based on the prefix CYP followed by an Arabic numeral indicating the protein family (usually >40% sequence identity), followed by a letter for the sub-family (>55% sequence identity), and the final number for the isozyme (often greater than 90% sequence identity). Importantly, genetic polymorphisms between CYP family members can result in differential processing of drugs between different genetic populations. For example, the drug diazepam (anxiety and seizure medication) is primarily metabolized by CYP3A4 and CYP2C19. However, mutations in 2% of Europeans, 14% of East Asians, and 57% of Oceanians result in deficient/absent CYP2C19 activity which leads to prolonged effects of the drugs in these patients. Poor drug metabolism can lead to an exaggerated pharmacological response, and the longer lifetime of the compound can also increase toxicity. Conversely, super-fast or rapid metabolizers (individuals with highly active CYP isoforms) can have altered drug responses by due to substantially lowered active drug lifespan. In both cases, clinical implications of polymorphisms require altering the dosages for the therapeutic regimen.
Since multiple drugs converge on CYP enzymes for metabolic processing, these oxygenases are also responsible for a number of drug-drug interactions. Several drugs are either substrates for CYP enzymes or act as inhibitors (prevent a substrate from engaging the CYP enzyme) or inducers (promote activity or gene synthesis of the CYP enzyme). For example, saquinavir (a protease inhibitor used for HIV treatment) is a substrate for CYP3A4, but can be administered with ritonavir (a CYP34A inhibitor) which enables a 33% increase in peak plasma concentrations of saquinavir. Similarly, serotonin uptake inhibitors such as fluoxetine inhibit CYP2D6. CYP2D6 processes codeine to morphine and a patient prescribed these inhibitors would therefore not benefit from the analgesic properties of codeine. There are also multiple inducers, for example CYP1A2 bio-transforms carcinogenic chemicals from cigarette smoke and is also induced by these agents in a positive feedback cycle. CYP profiling to determine which enzymes process a newly developed drug is important to determine any contradictions and metabolism concerns.
The mechanism for CYP oxidation is based on free radical chemistry that leverages different oxidation states of iron to facilitate electron transfer between the metal centre bound within a porphyrin ring called protoporphyrin IX (Figure 5.1). The overall process involves a drug (CYP substrate) with a potentially oxidizable bond (X-H) binding the low-spin Fe3+ (one unpaired electron S = ½, with a hexacoordinate metal centre including one water molecule) leading to a high-spin Fe3+ (five unpaired electrons S = 5/2, pentacoordinate with no water, and a shortened Fe-S bond , pulling the iron slightly below the plane of the heme). The drug substrate binding also triggers reduction of Fe3+ to Fe2+ with an electron originating from NADPH. The reduced Fe2+ has a high affinity for diatomic gasses such as O2 (and also CO) and transfers the electron to generate a superoxide Fe3+ complex. The Fe3+ ion picks up an additional electron (again originating from NADPH), which is also transferred to the superoxide anion to generate a peroxide anion (O22-) species that is split by two protons to form one water molecule and an iron-oxo complex called oxene. This oxene species is highly electrophilic and reactive and inserts into the substrate X-H bond yielding the desired oxidized product. This process only superficially describes the crystal field theory required for understanding these redox reactions, and the most relevant aspects for medicinal chemists are in understanding the types of C–H bonds that are susceptible to CYP oxidation and their final products.
FIGURE 5.1 Reaction mechanism for metabolism via CYP enzymes.
5.2.2 Oxidation of sp3 Carbon Atoms
5.2.2.1 Aliphatic Straight Chain and Ring Systems
The main mechanism for metabolism of an sp3 carbon atom is hydroxylation. If there are multiple sp3 carbon atoms in the drug, there is a preference that hydroxylation will occur at the more substituted carbon atom. This site selectivity arises from the electronics of radical stability. Consider the homolysis of the C-H bond during cleavage in Figure 5.2. The radical generated on the carbon atom is most stable on the ‘internal’ carbon atom and forms the major product. Regardless, the alcohol generated from both the major and minor products will be further oxidized by a non-CYP Phase I enzyme called Alcohol Dehydrogenase (ADH) to give a carbonyl (ketone or aldehyde). For any aldehydes generated as a result of terminal carbon oxidation, the molecule is further oxidized by a comparable Aldehyde Dehydrogenase (ALDH2) to yield a carboxylic acid. Notably, alcohol dehydrogenation is reversible, but the activity of aldehyde dehydrogenase is not reversible.
FIGURE 5.2 CYP-mediated oxidation of carbon centres preferentially occurs on the ‘internal’ carbon atom.
Saturated ring systems are a common functionality in different drug molecules. Due to sterics, oxidation generally occurs at the 3- or 4- ring position as shown with the metabolism of acetohexamide in Figure 5.3. Substitution at the 4- position during SAR design is often considered to impede Phase I enzyme access and sterically block hydroxylation, thereby extending drug half-life.
FIGURE 5.3 Acetohexamide is metabolized on the 4-position of the cyclohexane ring.
5.2.2.2 Allylic and Propargylic Systems
As shown with both examples above, both sterics and electronics can play a role in metabolism at sp3 carbon atoms. As such, there is a site preference of hydroxylation of sp3 carbon atoms alpha to an unsaturated system. Based on electronic effects, the proximity to a double/triple bond provides stability to a radical (Figure 5.4). Furthermore, in an unsaturated system, the resulting alcohol is dehydrogenated to create a 1,4-conjugated system which has excellent electrophilic properties for Phase II metabolism.
FIGURE 5.4 Allylic and propargylic carbons are more susceptible to oxidation.
Hexobarbital (a barbiturate derivative used as an anesthetic in the mid-20th century) contains a cyclohexene substituent (Figure 5.5). Based on sterics, metabolism could be favoured at the 4-position. However, hexobarbital is hydroxylated by CYP2B1 at the 3- position, alpha to the double bond (due to increased electronic stability of the radical). The resulting alcohol is either oxidized to an α,β-unsaturated ketone or is a substrate for UDP-glucuronosyltransferases (discussed in later sections on Phase II metabolism).
FIGURE 5.5 Hexobarbital is metabolized at C-3 position.
5.2.2.3 Halogenated Systems
In the case of a halogenated sp3 centre, oxidation (hydroxylation) can lead to a high-energy / unstable intermediate, that can undergo elimination of the halogen to form a ketone. This oxidative dehalogenation is more prevalent in the presence of two halogen atoms on the sp3 centre. For example, chloramphenicol is an antibiotic that disrupts bacterial protein synthesis. Chloramphenicol possesses a terminal di-chloro moiety that is oxidized to an acid halide intermediate (very reactive) and further reacts to form a terminal acid moiety. Note that oxidative dehalogenation is not feasible with fluorinated carbon atoms, as the C-F bond is extremely strong and cannot be homolyzed by CYP enzymes. In this way, fluorine can function as a bioisostere for hydrogen.
FIGURE 5.6 Oxidation of halogenated systems.
5.2.3 Oxidation of sp2 and sp Carbon Atoms
Unsaturated carbon scaffolds are also susceptible to oxidation by CYPs. For example, the oxygen atom from the CYP enzyme can insert into the π system of benzene to form a highly reactive epoxide (Figure. 5.7). The reactive epoxide is subsequently hydrolyzed by epoxide hydrolase to yield a dihydrodiol or it can re-arrange and re-aromatize to phenol via proton catalysis. In either case, these substrates are further oxidized to catechol (bis-phenol) derivatives. These catechols are substantially more polar and can be directly excreted, or oxidized by dehydrogenases to yield reactive quinones (which are substrates for Phase II metabolism).
FIGURE 5.7 Oxidation of benzene ring systems to form reactive quinones.
Acetanilide is a discontinued drug that was the first aniline derivative identified to possess analgesic properties. These favourable therapeutic properties are a result of aromatic hydroxylation at the para-position (sp2 carbon) which gives paracetamol (commonly known as acetaminophen or Tylenol). (Figure 5.8) However, there is also a minor metabolic product of N-dealkylation (discussed below) which results in aniline and is nephrotoxic leading to hemolytic anemias and other safety concerns.
FIGURE 5.8 Oxidation of acetanilide.
Compared to sp3 oxidation, the metabolic modification of sp2 and sp carbons is a significantly slower process. Furthermore, it is also uncommon in modern drug discovery to observe free, unsubstituted phenyl rings in drug structures. This is partly because common strategies for bypassing sp2 metabolism involve generating more electron deficient rings or adding a fluorine or chlorine/substituent to the phenyl ring.
The utility of benzene in synthetic organic chemistry represents an interesting example of differences in drug metabolism of sp3 and sp2 carbons. As a simple aromatic liquid, benzene was routinely employed as an organic solvent for a number of industrial applications as well as organic synthesis. However, it is highly toxic and has been largely superseded by toluene, which still has safety concerns, but is far less toxic than benzene. This difference arises from the sp3 benzylic position of toluene which is much weaker (C-H bond strength = 85 kcal/mol) than the aryl C-H bond (110 kcal/mol). As such the benzylic carbon is more readily oxidized by CYP enzymes, creating a benzylic radical that is further dehydrogenated and oxidized to afford benzoic acid, which is rapidly cleared. Conversely, oxidation of the benzene sp2 carbon is substantially slower, and therefore benzene remains in the body for longer time periods. Moreover, the toluene oxidation metabolite has limited safety challenges, whereas benzene oxidation leads to a reactive quinone electrophile which is an excellent 1,4-Michael acceptor.
5.2.4 Oxidation of Hetero-atoms Bound to Carbon Atoms
Hetero-atoms (O, N, and S) are common in drug discovery and are often responsible for key target-drug interactions. The general strategy employed by Phase I metabolism enzymes is to either i) oxidize the hetero-atom directly (referred to as heteroatom oxygenation) or ii) oxidize the alpha-carbon next to the hetero-atom (often referred to as de-alkylation although there is an initial hydroxylation step; Figure 5.9).
FIGURE 5.9 Oxidation of hetero-atoms proceeds either by SET (top) or HAT (bottom) mechanisms.
In heteroatom oxygenation, there is an initial single electron transfer (SET) from the heteroatom to the iron-oxygen complex of the CYP enzyme. This loss of an electron creates a radical cation intermediate on the heteroatom that eventually reacts with an oxygen provided by the CYP enzyme. In de-alkylation, there is an initial hydrogen atom abstraction (HAT) from the alpha-carbon which ends up on the iron-oxygen complex of the CYP enzyme (becoming iron-hydroxyl). This loss of a hydrogen atom creates a neutral carbon radical intermediate that eventually reacts with the hydroxyl provided by the CYP enzyme. The type of metabolism (heteroatom oxygenation or de-alkylation via SET or HAT respectively) that occurs depends on i) the hetero-atom and ii) the substituents of the heteroatom.
For carbon-oxygen systems, heteroatom oxygenation does not occur because removing a single electron to generate a positively charged oxygen is highly energetically unfavorable. Therefore, for carbon-oxygen systems, metabolism occurs via HAT and de-alkylation.
For carbon-nitrogen systems, HAT and dealkylation are highly favoured (similar to carbon-oxygen systems above). However, SET is not as unfavourable for nitrogen species and therefore heteroatom oxygenation is feasible and can proceed (usually as a minor product). SET/Hetero-atom oxidation is more favorable when the radical cation is destabilized based on the nearby substituents, or if there are no alpha-protons available for abstraction.
For carbon-sulfur systems, SET and heteroatom oxygenation is energetically favored due to the formation of a stable sulfur radical cation.
5.2.4.1 Metabolism of O-Containing Moieties
5.2.4.1.1 Metabolism via O-Dealkylation
As mentioned above, the carbon-oxygen containing moieties will only undergo de-alkylation and will not be directly oxidized on the oxygen atom. These C-O moieties are commonly found as ethers in drugs and will metabolize to an alcohol with release of a ketone or aldehyde. As with all metabolic reactions, steric hindrance of the ether can block metabolism. A well-known example of O-dealkylation involves metabolism of codeine to morphine where the terminal ether is metabolized and also releases the parent phenol (Figure 5.10). Notably, this only accounts for ~10% of the modification of ingested codeine. Approximately 10% of codeine is also N-dealkylated (which will be discussed in the next section), and the remaining 80% is directly glucuronidated on the cyclohexenyl alcohol, in Phase II metabolism. Notably, the alternative C-O linkage in the ring system is not metabolized due to steric hindrance.
FIGURE 5.10 Codeine is dealkylated to yield the analgesic morphine.
5.2.4.2 Metabolism of N-Containing Moieties
5.2.4.2.1 Metabolism via N-Dealkylation
The most common types of nitrogen containing compounds in drug discovery are tertiary amines (including both aliphatic and aromatic amines). Oxidation of these amines predominantly occurs as discussed above, where a carbon atom alpha to the nitrogen is hydroxylated to form a carbinolamine and this intermediate rearranges via cleavage of the C-N bond (de-alkylation; Figure 5.11).
FIGURE 5.11 Oxidation of secondary amines can lead to N-dealkylation.
For example, the tertiary amine of diazepam is N-dealkylated in Phase I metabolism (Figure 5.12). Additional metabolic steps of diazepam include hydroxylation of the sp3 carbon which becomes an available substrate for glucuronidation.
FIGURE 5.12 Oxidation route of diazepam.
Dealkylation occurs for tertiary and secondary amines. Primary aliphatic amines are metabolized via the same mechanism, where hydroxylation of the alpha-carbon occurs, followed by rearrangement to release ammonia, and this is termed de-amination (as opposed to N-dealkylation). Amphetamine is a respiratory and CNS stimulant that is metabolically processed by different reactions including an oxidative deamination but also β-hydroxylation (of the sp3 benzylic carbon) and para-hydroxylation (of the sp2 phenyl ring; Figure 5.13).
FIGURE 5.13 Differential metabolism routes of amphetamine.
5.2.4.2.2 Metabolism via N-Oxidation
In addition to de-alkylation and de-amination, tertiary amines can undergo metabolic N-oxidation by a group of Phase I enzymes called flavin-containing monooxygenases (FMO) which enable oxidation on the heteroatom to yield N-oxide products. As mentioned, this is generally less favourable due to initial formation of a radical cation on the nitrogen atom. For instance, imipramine is an amine-based tricyclic antidepressant. Its tertiary amine is predominantly processed by hepatocytes through N-dealkylation via CYP3A4 (84%). Para-hydroxylation by CYP2D6 accounts for ~10% of the metabolic products observed. Finally, the remaining 6% is metabolized to an imipramine-N-oxide via FMO (Figure 5.14).
FIGURE 5.14 Oxidation route of imipramine. Despramine can be further metabolized by additional N-dealkylation or or hydroxylation on the sp3 ring system.
N-Oxidation can also occur with alicyclic amines such as nicotine, which harbours a nitrogen in the pyrrolidine and pyridine rings. Approximately 10% of nicotine is N-dealkylated at the pyrrolidine and ~75% is oxidized to cotinine (amide) via a similar hydroxylation mechanism. The pyridine can also be directly glucuronidated through Phase II processes (5%). Additional metabolism occurs through FMOs to create nicotine n-oxide (Figure 5.15).
FIGURE 5.15 Nicotine can be metabolized at different carbon and nitrogen centres as described by Hukkanen et al (2005).
Nitrogen–containing aliphatic, and heteroaromatic ring systems are extremely common substructures in many drugs. Indoles and pyrroles represent electron-rich heterocycles which helps to facilitate metabolism via ring oxidation, with a preference towards oxidation on carbon atoms adjacent to the nitrogen. Metabolism trends from the analysis of several drug libraries indicate that a greater number of nitrogen atoms in the ring, reduce the possibility of Phase I oxidations. Tetrazoles in particular, are relatively inert to oxidation (and are important bio-isosteres of carboxylic acids, discussed later). Importantly, a number of nitrogen containing heterocycles including pyridines, imidazoles, and triazoles can engage with the heme-group to block activity of CYP enzymes. However, they can also be excellent substrates for Phase II metabolism, especially N-glucuronidation as observed with nicotine (Figure 5.16).
FIGURE 5.16 Oxidation of azole rings can break the aromaticity
5.2.4.3 Metabolism of S-Containing Moieties
5.2.4.3.1 Metabolism via S-Oxidation
Phase I metabolism for sulfur containing compounds generally proceeds via direct oxidation of the heteroatom (SET), which impacts different sulfur containing functional groups including sulfides (especially in ring structures), free thiols, sulfonyls, and thioketones. Free thiols are uncommon in modern drug molecules due to their reducing potential, which enables them to form di-sulfide bridges with cysteine residues or self-aggregate. However, in such drugs, the thiol is directly hydroxylated to form sulfenic acid, which is further oxidized to sulfonic acid (Figure 5.17).
FIGURE 5.17 Thiols are oxidized to sulfonic acids.
Higher oxidation states of sulfur, such as sulfonyls found in sulfonylureas and sulfonylamides, are significantly more hydrophilic, which facilitates their elimination. Thioethers (sulfides) can be oxidized to the corresponding sulfoxide and then further oxidized to the sulfone. For example, thioridazine (an antipsychotic medication used for schizophrenia) contains two sulfides and both of these sulfides can be S-oxidized (additional metabolic pathways include N-dealkylation; see Figure 5.18a). Thioketones undergo desulfuration via a similar oxidation of the sulfur atom as seen with thiopental, which produces a carbonyl-containing product (Figure 5.18b).
FIGURE 5.18 Metabolism of the sulfur-containing thioridazine as described by Wójcikowski et al (2006).
Importantly, metabolism of sulfur atoms has been associated with bioactivation and generation of reactive metabolites that are associated with toxicities, and understanding metabolite profiles can be important when selecting sulfur functional groups.
5.2.5 Other Phase I Oxidation Enzymes
In addition to CYPs, there are other non-CYP enzymes involved in Phase I metabolism, some of which have been highlighted above. The most commonly known non-CYP Phase I enzyme is Alcohol Dehydrogenase, which oxidizes alcohol functional groups into aldehydes or ketones, and Aldehyde Dehydrogenase, which oxidizes aldehydes to carboxylic acids. These enzymes are found exclusively in hepatocytes and have well characterized functions on ethanol, methanol, and ethylene glycol. Other important enzymes are Flavin Containing Monooxygenases (FMOs), which operate in a similar manner to CYPs and yield highly similar by-products. However, while CYPs utilize the high-reactive oxene as an oxidizing agent, FMOs use slightly less powerful peroxides. As such, FMOs can only oxidize heteroatoms and cannot oxidize primary amines, highly charged or polyvalent species.
Regardless of the oxidation mechanism, the overall goal of Phase I enzymes is to increase solubility (hydrophilicity) or expose a functional group for conjugation in Phase II.
5.2.6 Reductions
5.2.6.1 Carbonyls
Reductions are less common since they usually mask reactive groups, but they are largely involved in reducing carbonyls (mainly ketones) to hydroxy groups, which are more susceptible to conjugation reactions (Phase II). The order for reactivity generally follows the principles of sterics and electronics, namely aliphatic ketones/aldehyde > aromatic (benzylic) ketones/aldehyde > esters, acids, and amides. Importantly, the reduction of ketones can lead to the generation of stereocentres and stereoisomeric alcohols. For example, one of the routes for the metabolism of the blood thinner warfarin involves the reduction of its ketone by Human Hepatic Cytosolic Reductase to yield two diastereomeric alcohols.
5.2.6.2 Nitro- and Azo- Compounds
Nitro- reductions are performed by NADPH-dependent microsomal and soluble nitroreductases, whereas NADPH-dependent multicomponent hepatic microsomal reductase systems reduce azo- groups. The end products of these reactions are primary amines (Figure 5.19). Bacterial reductases in the gastrointestinal tract have the capacity to reduce both nitro and azo groups.
FIGURE 5.19 Reductions of nitro- and azo- compounds can yield primary amines.
5.2.7 Hydrolysis
Hydrolysis reactions are fundamentally distinct from previous Phase I reactions described above, in that there is no change to the redox level of the drug molecule, and there is usually a substantial change to original substrate (cleavage of a functional group and generation of multiple products). The reactivity of a substrate towards hydrolysis is determined by the electrophilicity of the carbonyl carbon (or other reactive centre) and the nucleofugality of the leaving group substituents. The rate is found to follow the trend of thioester > ester > carbonate > amide > carbamate. One of the classic examples is hydrolysis of aspirin which can be converted to salicylic acid by esterases. (Figure 5.20)
FIGURE 5.20 Aspirin can be hydrolyzed to salicylic acid.
5.3 Phase II Metabolism
Whereas Phase I is largely focussed on oxidases that aim to introduce a polar functional group, Phase II involves transferases that conjugate a biomolecule onto the drug. Phase I reactions may not necessarily generate a pharmacologically inactive species, but Phase II reactions are regarded as detoxifying and will inactivate drugs to prevent damage to macromolecules or to prepare drugs for efficient elimination. Substrates are covalently coupled to an endogenous molecule, which is generally very polar and large (100-300 Da) and can usurp the physical properties of the drug (LogP, tPSA, etc.). Since these processes can be energetically unfavourable, Phase II transferase enzymes will activate the endogenous molecule to prepare it for conjugation to the drug.
5.3.1 UDP-Glucuronosyltransferases (UGT)
Glucuronidation by UGTs represents the most common Phase II metabolic process and involves the transfer of a glucuronosyl group to either an O-, N-, S– or C– atom. Physiologically, the ubiquity of glucuronidation arises from the availability of D-glucuronic acid (a D-glucose derivative), the enzymatic activation of the conjugate, and the stark changes in water solubility by addition of the glucose-group. In this conjugation reaction, glucose-1-phosphate is first activated to uridine-5-diphosphoate-alpha-glucose (UDPG) by the action of a phosphorylase. The 6-C is oxidized to an acid via UDPG-dehydrogenase and two equivalents of NAD+ (Figure 5.21a). The glycosidic bond is a high energy bond and easily broken to conjugate onto the substrate via nucleophilic attack of the drug substrate. This results in uridine diphosphate (UDP) functioning as a leaving group. Notably, this SN2 attack of the drug substrate also leads to inversion of stereochemical configuration in the resulting β-linkage to the glucuronic acid. A number of functional groups can participate in this reaction, including weak nucleophiles, and this accounts for approximately one-third of all Phase II metabolism. It is very common to form O-glucuronides via a hydroxy groups (phenolic/alcoholic) or carboxylates (aromatic or aliphatic). For example, morphine is extensively metabolized via O-glucuronidation (Figure 5.21b).
Even carbon atoms, in a minority of cases, can be glucuronidated such as in the case of sulfinpyrazone (a drug for the chronic treatment of gout). This occurs because the H-atom of the tertiary carbon is acidic and susceptible to removal by a base to create a nucleophilic carbanion that undergoes reaction with UDP-glucuronic acid (UDPGA) (Figure 5.21c). As described in previous examples, N-atoms and S-atoms can also be glucuronidated.
FIGURE 5.21 Generation of the substrate and mechanism of conjugation for glucuronidation.
5.3.2 Glutathione-S-Transferase
Glutathionylation is the second-most common Phase II metabolism pathway, and also forms a significant aspect of first-pass metabolism. Glutathionylation is thought of as a highly protective process with de-toxifying effects. Glutathione is a tri-peptide analogue of glycine, cysteine, and glutamate, with a γ-peptide linkage between the carboxyl side chain of glutamate and the alpha amino group of cysteine. The reactivity of glutathione emerges from the cysteine side chain, which has an expected pKa of 8.7 (more acidic than a similar alcohol functionality such as serine, due to stabilization of the negative charge on a larger, polarizable sulfur atom). However, the nucleophilic character can be further enhanced by the action of the enzyme Glutathione S-transferase, which can increase the acidity by 2-3 pKa units, generating a charged thiolate anion. Any drug that has mildly electrophilic sites will be vulnerable to nucleophilic attack. The most common substrates of glutathione-S-transferases are soft electrophiles/Michael acceptors.
α,β-unsaturated carbonyls (most common electrophile)
quinones/quinone imine (often formed on Phase I metabolism)
epoxides FIGURE 5.22 The tri-peptide glutathione.
There have been a number of drug examples discussed above that are subject to glutathionylation, such as paracetamol (Tylenol). One case involves remoxipride, which is a substituted benzamide and was previously employed as an antipsychotic for treatment of schizophrenia. As expected, initial CYP metabolism leads to dealkylation of an ether linkage that is further oxidized to a quinone species (Figure 5.23). The presence of a bromo-group enables two glutathione transfer reactions, which inactivates the molecule and prepares it for elimination. Remoxipride was discontinued after 8 cases (2 fatal) of aplastic anemia were detected.
FIGURE 5.23 Gluathionation of remoxipride following oxidation of substituents on the benzene ring.
5.3.3 Sulfonyltransferases
Sulfonylation involves the transfer of sulfate from an endogenous biomolecule to a drug (containing a nucleophilic hetero-atom). Similar to the above Phase II reactions, the introduction of a sulfate markedly alters the hydrophilicity. The sulfate group is provided by a molecule called 3′-phosphoadenosine 5′-phosphosulfate (PAPS) which contains a mixed sulfate/phosphate anhydride. This is a high energy anhydride that is significantly more reactive than di- or tri-phosphates, but has significantly reduced cellular availability compared to glutathione or UDP-glucose. This reaction predominantly occurs with phenols, but is known to occur with alcohols, aryl amines, and N-oxide compounds. For example, minoxidil (Rogaine) is drug used to help with hair loss in men and can be metabolized by the action of sulfonyltransferases (as well as by glucuronidation; Figure 5.24).
FIGURE 5.24 Minoxidil can be metabolized via oxidation of the glucuronidation or sulfonylation as described by Kikuchi et al. (2016).
5.3.4 Methyl Transferases
While glucuronidation, glutathionation, sulfonylation, as well as several other Phase II metabolism pathways increase the water-solubility of drugs, processes such as methylation (and acetylation below) do not substantially alter values such as LogP. Instead, these processes are tailored more toward inactivating a drug (an exception to his occurs with the formation of a charged, quaternary ammonium derivative). Any heteroatom with a hydrogen atom is prone to methylation, and this usually occurs at a late stage in the metabolism process. Methylation is carried out by S-adenosylmethionine (SAM) which involves an ATP-activated methyl group from methionine). For example, theophylline is used to treat apnea and breathing issues (which can be common in infants, especially premature babies), but the methylated metabolite is caffeine, which can lead to adverse effects (Figure 5.25).
FIGURE 5.25 Methyl groups can be activated by methionine adenosyl transferase, and added on to specific hetero-atoms such as in the generation of caffeine.
5.3.5 Acetyl Transferases
Similar to methylation, acetylation does not substantially alter lipophilicity but facilitates drug inactivation particularly on nucleophilic atoms (N and O). The main substrates are arylamines and sulfonamides, and to a lesser extent hydrazines, hydradizes, and aliphatic amines. The major enzyme system that acetylates the drugs is arylamine N-acetyltransferase. Primary and secondary aliphatic amines are only marginally acetylated (Figure 5.26). This reaction is particularly important in sulfonamide metabolism because acetyl-sulfonamides have reduced solubility and can lead to precipitation, especially in the kidneys, leading to renal toxicities
FIGURE 5.26 Routes for acetylation of nitrogen-containing moieties.
5.4 Drug Metabolism Exploits Ubiquitous Themes in Chemistry
One of the remarkable features of hepatocytes and drug metabolism within the body is that there is a high efficiency and capacity to metabolize foreign molecules with a wide range of chemical diversity. Instead of developing a tailored nucleophile to react with specific drug, general mechanisms are used to oxidize generic scaffolds found in all drug molecules. This greatly simplifies the extent of biological tools needed and improves genetic efficiency for drug metabolism while maintaining a broad reactivity and substrate scope.
Furthermore, not every single oxidation, reduction, hydration, etc., that can occur will occur. The above examples outline potential reactions with sp3 and sp2 carbons, heteroatoms, acids, and bases. Many drugs contain multiple functional groups, and there is usually a preference for specific metabolite profiles that depends on multiple features including the steric hinderance, electrophilicity, and polarity of the molecule. Once the molecule undergoes an initial modification, it may be sufficiently water-soluble to be expelled without further modification. However, in the same way, a single modification may not be sufficient and there can be multiple modifications that occur.
A key part of medicinal chemistry programs is identifying the metabolic liabilities on the drug – positions that are metabolized. This can help extend the lifetime of the drug or identify Phase I and Phase II metabolites. A metabolite identification (MetID) experiment is performed where a compound is subjected to hepatocytes and the resulting products are identified through mass spectrometry. The resulting pathway may not be linear and may demonstrate different transformations occurring at the different stages although they all converge on the common goal of preparing the compound for elimination.
Drug Modifications to Improve Stability
6
“You can’t use up creativity. The more you use, the more you have.”
– Maya Angelou
6.1 Isosteres and Bioisosteres
Improving ADME properties is a fundamental aspect of medicinal chemistry to limit the effects of first-pass metabolism while maximizing the efficacy of a drug candidate for its target. The general strategy to accomplish this is to employ isosteres or bioisoteres in a lead or drug candidate. While isomers refer to molecules that have the same number and type of atoms (albeit in different three-dimensional arrangements), isosteres refer to molecules that have different atoms but similar electronic properties. Likewise, bioisosteres have different atoms (which can lead to different chemical or physical properties) but share similar electronics or geometry, which results in the capacity of offering a similar biological function as the original ligand.
Throughout hit-to-lead and lead optimization, identifying bioisosteres that have functional mimicry is a key aspect of designing an SAR study. For example, replacing a thiol with a hydroxyl group preserves the use of an electronegative atom from the same group of the periodic table while offering an HBD. Some bioisosteres maybe non-obvious, for example, a catechol can be mimicked by a benzimidazole. This is because the neighbouring hydroxyl groups of the bis-phenol can participate in hydrogen bonding, which leads to a pseudo-five membered ring with a free hydrogen. In this way, the benzimidazole geometrically mimics this structure and contains a free hydrogen bond donor (Figure 6.1).
FIGURE 6.1 Re-drawing interactions between functional groups can reveal different bioisosteres.
Over the past century, exploration of chemical space across hundreds of drugs has led to a ‘dictionary’ of bioisosteres that can be employed and evaluated in drug design. There are generally two types: classical and non-classical bioisosteres. Classical bioisosteres are structural analogs and are usually readily identifiable because they have similar sizes. Classical bioisosteres can be catalogued as monovalent/univalent (eg. CH3 ~ NH2 ~ OH ~ F ~ Cl), bivalent (eg. vinyl ~ carbonyl ~ imine ~ thioketone), trivalent (eg. alkynyl ~ nitrile), tetrasubstituted (tetrasubstituted carbon ~ tetrasubstituted nitrogen) and ring equivalent (eg. benzene ~ pyridine ~ thiophene). Non-classical analogues aim to mimic either the geometry or electronics/H-bonding capacity, but may look structurally distinct from the functional group being imitated. (eg. carboxylic acid ~ tetrazole). In either case, bioisosteres represent a strategy to preserve specific target-molecule interactions while modulating lipophilicity, solubility, acidity/basicity, and other properties.
6.2 Bioisosteres of Hydrogen
Depending on their location within a molecule, hydrogen atoms can sometimes be a metabolic soft spot. There are two main bioisosteres for hydrogen atoms. The most similar isostere is deuterium, a heavy hydrogen isotope (and is also non-radioactive and naturally occurring). As an isotope, the chemical properties of hydrogen are essentially preserved in replacement with deuterium. However, the additional neutron doubles the atomic mass which drastically alters the kinetics of a reaction (deuterium kinetic isotope effect). This becomes important if the hydrogen is involved in a catalytic step or as a hydrogen bond donor. Although they are naturally occurring, deuterium isotopes have a significantly increased costs associated with synthesis, and are not frequently employed as bioisosteres. Deucravacitinib is a selective Janus kinase (JAK) inhibitor of TYK2 that contains a magic-methyl moiety that is critical for activity (Figure 6.2). This methyl (located on the amide) is deuterated which blocks metabolism of the sp3 site (the C-D bond is ~5-10-fold stronger than the C-H bond due to lower vibration frequencies). Deuterium is the only isotope commonly considered in medicinal chemistry because the mass change is not as significant for larger atoms to substantially alter reaction kinetics (although other atomic isotopes may be employed in drugs for purposes such as imaging or radiation therapy).
FIGURE 6.2 Deucravacitinib contains a deuterated methyl group that is essential for bio-activity.
A more commonly employed isostere for hydrogen is fluorine. This may seem unusual as fluorine is a highly electronegative halogen. However, the electronegativity of fluorine reduces the size of the electron cloud leading to similar sizes (carbon bond lengths of 1.20 Å [C-H] vs 1.35 Å [C-F]). The electronegativity difference makes the carbon-fluorine bond the strongest bond in organic chemistry because of the partial ionic character of the bond. This makes C-F groups highly resistant to oxidation by CYP enzymes. For example, the cholesterol absorption inhibitor, ezetimibe, was generated with fluorine atoms at the para-positions on both free benzene rings to reduce the metabolic instability of the para-hydrogen atoms (Figure 6.3). Importantly, the electron-withdrawing inductive effects of fluorine should also be considered when substituting a fluorine into a molecule (for example the impact on a nearby HBA).
FIGURE 6.3 Protecting the para-position of free benzene rings can improve metabolic stability and lower effective doses.
6.3 Bioisosteres of Phenyl Rings
Phenyl rings are extremely common in drug discovery and possess a planar structure and engage in multiple hydrophobic interactions. Classical bioisosteres of phenyl rings aim to preserve the aromaticity and/or planar structure such as the previously described example of Viagra to Levitra involved a straightforward substitution of a carbon atom for a nitrogen atom (Figure 6.4a). Other examples include the SAR study performed by Liang et al (2016) and summarized by Subbaiah et al (2021), these hetero-atom substitutions on a phenyl ring in KDM5 inhibitors can facilitate new interactions and provide a scaffold for additional substitutions (Figure 6.4b).
FIGURE 6.4 Potential bioisosteres for benzene rings that leverage hetero-atoms in ring systems.
There are less obvious bioisosteres for phenyl rings where the goal is to preserve the exit vector of a substituent. For example, cyclopropane can be employed as a bioisostere in some cases. In Factor Xa inhibitors generated by Brystol Myers Squibb, replacement of the phenyl ring with a cyclopropyl ring maintained the orientation of the tertiary amine, but substantially reduced the molecular weight thus increasing ligand efficiency (Figure 6.5). In this case, the phenyl ring was not functionalized, and the efficacy of these substitutions depends on the chemical species that can be accommodated in the pocket as well as the resulting changes to LogP / lipophilicity.
FIGURE 6.5 Cyclopropyl groups can serve as a benzene isosteres due to the preserved geometry of the substituents.
Other non-classical bioisosteres of phenyl rings that have emerged include saturated bi- and polycyclic-rings such as cubanes, bi-cyclopentanes, and bi-cyclohexanes. These replacements are known to increase both metabolic stability (reduced 1,4-quinone formation) and water solubility (reduced π-π stacking interactions) while preserving dihedral angles (180°). These polycyclic rings show optimal functional mimicry when the original role of the phenyl ring is as a spacer (as opposed to participating in key interactions such as aryl-protein π-π stacking). For example, replacement of the phenyl ring with a bicyclohexyl group in the MDM2 inhibitor was shown to maintain potency but with greatly increased metabolic stability leading to a 6-fold increase in PK exposure (Figure 6.6). For these substitutions it is important to consider the linker length in deciding which bi-cyclo ring to install. For example, the end-to-end distance of C1-C4 atoms of benzene is 2.82 Å, while the comparable distance in a bi-cyclohexane, bi-cyclopentane, and bi-cyclobutane is 2.72 Å, 1.70 Å, and 2.60 Å respectively. Incorporation of these (and other) polycyclic rings can potentially enable precise adjustment of linker lengths.
FIGURE 6.6 Bridged ring systems can serve as bio-isosteres for benzene rings and reduce the overall planarity while maintaining potency.
6.4 Bioisosteres of Carboxylic Acids
Bioisosteres for carboxylic acids have been heavily explored because of the anionic nature of the functional group which exhibits large (and usually unfavourable) effects on permeability and PK. The negative charge of the carboxylate group is delocalized over two oxygen atoms and responsible for potency with many protein targets via ionic interactions. Therefore, bioisosteres usually require an acidic proton as well as a two-point interaction profile to function as a suitable surrogate. The most common (non-classical) bio-isostere is a tetrazole, which contains an acidic proton (pKa ~ 4.5) but is ~10-fold more lipophilic. For example, potent inhibitors of AT1 (GPCR) receptor were developed based on a carboxylic acid-receptor interaction but only available by IV injection (Figure 6.7a). Replacement with a tetrazole improved binding but also enabled oral administration due to increased lipophilicity and led to the antihypertensive drug, losartan. There are hundreds of different types of carboxylic acid bioisosteres that have been explored, such as difluorophenols, squaric acids, and hydroxyquinoline-2-ones (Figure 6.7b).
FIGURE 6.7 Examples of bio-isosteres for carboxylic acids.
6,5 Bioisosteres of Amides
Amide bonds may be metabolically unstable in drugs due to their enzymatic sensitivity towards proteases (often targeted towards peptide bonds). Bioisosteres of amides are designed to mimic the polarity as well as the planar geometry of the amide bond. Functional groups such as ketones, carbamates, ureas, and esters, can all mimic the polarity of the amide bond as well as the geometry. Triazoles can mimic both the H-bond acceptor and H-bond donor properties while maintaining similar spacer distances. In a similar manner, oxazoles, imidazoles, pyrazoles, thizaoles, can also substitute as cyclic replacements for an amide. Therefore, there are multiple functionalities that can serve as appropriate substituents, which provide opportunities for custom tailoring.
Less conservative changes have more direct atom substitutions but attempt to preserve the geometry and HBD and HBA character. For example, trifluorethylamine can be used to replace the amide bond in the Cathepsin K inhibitors developed by Merck (Figure 6.8). This allows for reducing the basicity of the amine while still enabling HBD properties and preserving the 120° bond geometry and polarity (the C-CF3 bond has been determined to be as polar as the carbonyl C=O bond).
FIGURE 6.8 Installation of a bio-isosteres for the amide functionalities can improve potency and stability as demonstrated by Cathepsin K inhibitors.
Oxetanes are also potential replacements for the carbonyl of an amide since they are stable and are significantly polar. The positioning of the oxygen within a 4-membered ring results in a larger space for the lone-oxygen pairs, potentially improving their role as HBAs. Oxetanes can also be used as bioisosteres for gem-dimethyl groups where they adopt similar steric volumes without the lipophilicity increases of the methyl substituents. These functional groups also have increased sp3 character with reduced susceptibility to CYP oxidation.
An alternative strategy involves transposing the amide, which increases resistance to protease activity (metabolic stability) and can have limited effects on target engagement, H-bonding, and conformational mobility. For example, transposition of the amide in the bacterial AKR1C3 inhibitor substantially improved compound selectivity through improved HBD contacts.
FIGURE 6.9 In certain cases, transposing an amide can maintain biological activity while improving other features such as isoform selectivity or metabolic stability.
6.6 Overall Approach to Bio-isosterism
In general, the main goal for bio-isosterism involves recognizing metabolic or permeability soft-spots in the molecule and identifying appropriate functional groups that can serve as a replacement. This can involve more sophisticated approaches that are not simply a one-for-one substitution. For example, Astra Zeneca was generating cathepsin K inhibitors for treatment of osteoporosis and other bone related indications. Their lead compound, balicatib, was shown to be potent, leveraging interactions of the cyclohexyl bisamide moiety, although it had poor PK properties and was susceptible to glutathionation. (Figure 6.10a) Structural data suggested that modifications to the phenylpiperazine rings would not disrupt the core pharmacophore. Several bioisosteres were attempted to mimic the capped-bisphenol, which was found to be susceptible to oxidation and glutathione reactivity. For example, tying back the O-methoxy groups into a 5-ring structure resulted in additional Phase I metabolism. This was similar to the cyclopropyl group which is a comparable isostere for an oxygen atom (in size). Installing 5- or 6- membered rings with heteroatoms creates unique positions for the lone-pairs and electron density, with the indazole demonstrating the best overall properties. (Figure 6.10b)
FIGURE 6.10 Examples of the SAR for cathepsin K inhibitors and the use of bio-isosteres.
6. 7 Structural Alerts
There are literally hundreds of bioisosteres that can be employed in drug discovery and being able to recognize and suggest functional groups is a key part of the design process. Equally, identifying functional groups that can lead to toxicity or reactivity is a critical aspect. These functional groups are referred to as structural alerts on a molecule and can indicate potential mutagenicity, hepatic toxicity, renal toxicity, phospholipidosis, etc. Importantly, these functional groups can be structural alerts for toxicity in one drug, but not another, because of competing factors such as sterics or electronics that may block metabolic bioactivation.
6.7.1 Aromatic N-H and O-H
Aromatic N-H groups may be susceptible to nucleophilic attack, which generates a reactive nitrenium ion that can react with important biomolecules such as DNA resulting in mutagenic toxicities. (Figure 6.11a) Replacement of the aromatic amine can remove the structural alert such as substituting a methyl group on carbutamide to give tolbutamide (sulfonylureas for hyperglycemia). (Figure 6.11b) Non-aromatic amines can also be oxidized to form nitroso groups which are toxic metabolites (and therefore structural alerts). Similarly, nitro- groups are poor HBA and readily oxidized to nitroso-groups and often avoided in drug discovery. (Figure 6.11c)
FIGURE 6.11 Challenges with primary amines in drug discovery.
Similar to primary aromatic amines, aromatic alcohols are also structural alerts due to the formation of quinones, which are 1,4 Michael acceptors. These species are highly electrophilic and can react with nucleophilic biomolecules, leading to covalent attachment and irreversible damage. (Figure 6.12)
FIGURE 6.12 Phenolic oxygens are structural alerts on certain molecules.
6.7.2 Unsubstituted Rings
Structural alerts can also apply to aromatic and aliphatic ring systems, where unsubstituted rings are often a key indicator of potential metabolism. Cyclohexyl groups are often oxidized at the 4-position initially, and a common protection strategy is to block this position with a bioisostere such as fluorine. Similarly, changing a piperidine ring for a morpholine ring can offer solubility advantages while blocking the metabolic soft spot. Additional changes can also be feasible such as introducing ring strain or sp2 character. Likewise, unsubstituted benzene rings are structural alerts as these are subject to oxidation via epoxidation. The most common strategy is to deactivate the ring with an electron-withdrawing group, particularly at the para- position. Introduction of hetero-atoms in the ring can also be helpful, and the more nitrogen atoms present in the ring, the more resistant they are to CYP-mediated oxidation.
FIGURE 6.13 Unsubstituted rings can lead to metabolic liabilities.
6.7.3 Furans, Thiophenes, and Thiazoles
Five-membered hetero-substituted rings can also be structural alerts and can lead to protein or biomolecule covalent modification. (Figure 6.14a,b) Furans are substrates for oxidation (similar to benzene rings), which can lead to hydrolysis and generation of highly electrophilic species. Thiophenes can also lead to epoxidation but result in free thiols that lead to hepatotoxicity. Importantly, some of these structural alerts are dormant in different molecules. For example, the thiophene moiety is dormant in both canagliflozin (a sodium glucose co-transporter inhibitor) and rivaroxaban (a Factor Xa inhibitor). For canagliflozin, metabolism predominantly occurs via UGT enzymes on the glucose moiety. For the rivaroxaban, metabolism occurs on the morpholinone ring system. Both examples indicate that if there is steric hindrance or other sites that are more susceptible to oxidation, this can bypass potential structural alerts on a molecule. (Figure 6.14c)
FIGURE 6.14 Metabolism of furans and thiophenes can lead to covalent modification of different biomolecules as described by Dang et al (2017).
6.8 PAINS Molecules
In addition to recognizing structural alerts to toxicity, there are additional classes of Pan-Assay INterference compounds which are referred to as PAINs alerts. These are functional groups that often yield false positive hits in assays. PAINs molecules are important to recognize early-on in the hit identification process, because if they are advanced further in the drug discovery pipeline, significant resources can be invested into a non-binding compound. Furthermore, since these molecules may exhibit non-specific binding to the target, they cannot be optimized to improve binding. For instance, a quinone is highly reactive and can react with a protein non-specifically, disrupting its secondary structure. In an activity assay, this loss of secondary structure could be observed as a loss of activity, which would be similar to what would be observed if a molecule bound to the protein as a true hit. PAINs molecules become particularly important when a high-throughput screens of thousands of compounds are performed.
There are several recognized molecular scaffolds for PAINs activity. However, sometimes a molecule exhibits PAINs activity in a screen against a specific target but is an active-binding participant in the pharmacophore of another molecule. For example, if the scaffold has metal-chelating potential (such as a free-thiol), it could show PAINs activity against an array of metalloproteins but not against non-metalloproteins. Therefore, every drug program needs to evaluate PAINs moieties independently and early-on in the process. Orthogonal binding and activity assays are important to confirm hits, before embarking on a full SAR campaign.
An Outlook on Medicinal Chemistry
7
“Art is never finished, only abandoned.”
– Leonardo Da Vinci
7.1 Modern Medicinal Chemistry
Modern medicinal chemistry is a highly integrative field of science, that embodies nearly all disciplines of physical science in the effort to develop new pharmaceutical agents. Completion of the Human Genome Project in 2003 serves a launch-point for the vast expansion of multi-omics datasets, which, coupled with advances in computational power has begun to open a new era in in drug discovery, driven by collaborative workflow models and information networks. Advances in chemical biology, organic synthesis, and structural biophysics have enabled medicinal chemists to achieve creative, precise molecular intervention at the cellular level. Significant investment in data collection technologies (e.g. mass spectrometry proteomics) and data analysis strategies (bioinformatics/machine learning) have in some ways, “flipped” drug discovery workflows, where compound libraries are screened agnostically against any particular protein/enzyme, often leading to valuable new insights about previously “undruggable” targets in disease-driving pathways. Ultimately, all stages of the traditional pipeline must work in concert in order to bring transformative treatment options to patients and combat emerging disease-strains that are resistant to previous therapies.
7.2 New Technologies are Constantly Created
The integrative workflows of modern medicinal chemistry are back-dropped against a constantly growing toolbox of therapeutic technologies. The creativity of researchers in the field has been on full display over the last two decades, where a vast number of new mechanisms for modulating cellular proteins are emerging. A harmony between organic chemistry and chemical biology, among other disciplines, has engendered breakthroughs that go beyond the traditional ligand/enzyme binding equilibrium model in order to regulate target activity. For instance, the emergence of covalent inhibitors has changed the way medicinal chemists think about the classical concepts of potencies and occupancy, allowing more favourable target inhibition. Such developments are driving research in many challenging areas, such as targeting difficult or previously undruggable sites, such as those of protein-protein interactions. Alternatively, heterobifunctional ligands have recently undergone a massive stage of growth and development, and these technologies are now reaching the point of clinical evaluation. These bivalent molecules employ a clever mechanism of binding with two distinct cellular proteins simultaneously, with a chemical linker connecting the events. Chemists have now demonstrated this concept as a viable approach to affect a wide variety of post-translation modifications (e.g. phosphorylation) through proximity-induction between the target of interest and modifying enzyme. In concert with the rapidly expanding fields of proteomics, metabolomics, and computational biology, researchers are pushing the boundaries of the conventional archetypes in small molecule drug discovery. Guided by the well-established principles of molecular recognition and protein-ligand binding models used by medicinal chemists for over half a century, molecules of increasing complexity in cellular mechanism-of-action are being designed and validated. In the push to rectify high clinical attrition rates in drug discovery and achieve patient specific therapy, these technologies which depart from the traditional mechanisms for target modulation may provide the solution.
References and Resources
8
A, Tripathi, and Bankaitis VA. 2018. “Molecular Docking: From Lock and Key to Combination Lock.” Journal of Molecular Medicine and Clinical Applications 2(1).
Åberg, Veronica et al. 2008. “Carboxylic Acid Isosteres Improve the Activity of Ring-Fused 2-Pyridones That Inhibit Pilus Biogenesis in E. Coli.” Bioorganic and Medicinal Chemistry Letters 18(12): 3536–40.
Aguilar, Angelo et al. 2017. “Discovery of 4-((3′R,4′S,5′R)-6-Chloro-4′-(3-Chloro-2-Fluorophenyl)-1′-Ethyl-2-Oxodispiro[cyclohexane-1,2′-Pyrrolidine-3′,3-Indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-Carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development.” Journal of Medicinal Chemistry 60(7): 2819–39.
Alberts, David S. et al. 1996. “Intraperitoneal Cisplatin plus Intravenous Cyclophosphamide versus Intravenous Cisplatin plus Intravenous Cyclophosphamide for Stage III Ovarian Cancer.” New England Journal of Medicine 335(26): 1950–55.
Amano, Yasushi, Tomohiko Yamaguchi, and Eiki Tanabe. 2014. “Structural Insights into Binding of Inhibitors to Soluble Epoxide Hydrolase Gained by Fragment Screening and X-Ray Crystallography.” Bioorganic and Medicinal Chemistry 22(8): 2427–34.
de Araujo, Elvin D. et al. 2019. “Structural and Functional Consequences of the STAT5BN642H Driver Mutation.” Nature Communications 10(1).
de Araujo, Elvin D., György M. Keserű, Patrick T. Gunning, and Richard Moriggl. 2020. “Targeting STAT3 and STAT5 in Cancer.” Cancers 12(8): 1–8.
Attwood, Misty M. et al. 2021. “Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design.” Nature Reviews Drug Discovery 20(11): 839–61.
Ballatore, Carlo, Donna M. Huryn, and Amos B. Smith. 2013. “Carboxylic Acid (Bio)Isosteres in Drug Design.” ChemMedChem 8(3): 385–95.
Bennett, Joan W., and King Thom Chung. 2001. “Alexander Fleming and the Discovery of Penicillin.” Advances in Applied Microbiology 49: 163–84.
Bordon, Yvonne. 2016. “The Many Sides of Paul Ehrlich.” Nature Immunology 17(S1): S6–S6.
Bosch, Fèlix, and Laia Rosich. 2008. “The Contributions of Paul Ehrlich to Pharmacology: A Tribute on the Occasion of the Centenary of His Nobel Prize.” Pharmacology 82(3): 171–79.
Bregante, Javier et al. 2021. “Efficacy and Synergy of Small Molecule Inhibitors Targeting FLT3-ITD+ Acute Myeloid Leukemia.” Cancers 13(24).
Brook, Karolina, Jessica Bennett, and Sukumar P. Desai. 2017. “The Chemical History of Morphine: An 8000-Year Journey, from Resin to de-Novo Synthesis.” Journal of Anesthesia History 3(2): 50–55.
Carr, Robin A.E., Miles Congreve, Christopher W. Murray, and David C. Rees. 2005. “Fragment-Based Lead Discovery: Leads by Design.” Drug Discovery Today 10(14): 987–92.
Chalmers, Benjamin A. et al. 2016. “Validating Eaton’s Hypothesis: Cubane as a Benzene Bioisostere.” Angewandte Chemie – International Edition 55(11): 3580–85.
Cho, Naoki et al. 2017. “Identification of Novel Glutathione Adducts of Benzbromarone in Human Liver Microsomes.” Drug Metabolism and Pharmacokinetics 32(1): 46–52.
Clader, John W. 2004. “The Discovery of Ezetimibe: A View from Outside the Receptor.” Journal of Medicinal Chemistry 47(1): 1–9.
Cohen, Philip, Darren Cross, and Pasi A. Jänne. 2021. “Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions.” Nature Reviews Drug Discovery 20(7): 551–69.
Conner, Kip P., Caleb M. Woods, and William M. Atkins. 2011. “Interactions of Cytochrome P450s with Their Ligands.” Archives of Biochemistry and Biophysics 507(1): 56–65.
Crawford, James J. et al. 2012. “Pharmacokinetic Benefits of 3,4-Dimethoxy Substitution of a Phenyl Ring and Design of Isosteres Yielding Orally Available Cathepsin K Inhibitors.” Journal of Medicinal Chemistry 55(20): 8827–37.
Currie, Geoffrey M. 2018. “Pharmacology, Part 2: Introduction to Pharmacokinetics.” Journal of Nuclear Medicine Technology 46(3): 221–30.
Dalvie, Deepak K. et al. 2002. “Biotransformation Reactions of Five-Membered Aromatic Heterocyclic Rings.” Chemical Research in Toxicology 15(3): 269–99.
Dalvie, Deepak K., and Bernard Testa. 2021. “Principles of Drug Metabolism.” In Burger’s Medicinal Chemistry and Drug Discovery, , 1–139.
Dang, Na Le, Tyler B. Hughes, Grover P. Miller, and S. Joshua Swamidass. 2017. “Computational Approach to Structural Alerts: Furans, Phenols, Nitroaromatics, and Thiophenes.” Chemical Research in Toxicology 30(4): 1046–59.
Desborough, Michael J.R., and David M. Keeling. 2017. “The Aspirin Story – from Willow to Wonder Drug.” British Journal of Haematology 177(5): 674–83.
Dzobo, Kevin. 2022. “The Role of Natural Products as Sources of Therapeutic Agents for Innovative Drug Discovery.” In Comprehensive Pharmacology, , 408–22.
Eh-Haj, Babiker M. 2021. “Metabolic N-Dealkylation and N-Oxidation as Elucidators of the Role of Alkylamino Moieties in Drugs Acting at Various Receptors.” Molecules 26(7).
Elumalai, Nagarajan et al. 2015. “Nanomolar Inhibitors of the Transcription Factor STAT5b with High Selectivity over STAT5a.” Angewandte Chemie – International Edition 54(16): 4758–63.
Elumalai, Nagarajan et al. 2017. “Rational Development of Stafib-2: A Selective, Nanomolar Inhibitor of the Transcription Factor STAT5b.” Scientific Reports 7(1).
Feng, Minghao, Bingqing Tang, Steven H. Liang, and Xuefeng Jiang. 2016. “Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry.” Current Topics in Medicinal Chemistry 16(11): 1200–1216.
Ferreira, Leonardo G., and Adriano D. Andricopulo. 2017. “From Protein Structure to Small-Molecules: Recent Advances and Applications to Fragment-Based Drug Discovery.” Current Topics in Medicinal Chemistry 17(20).
Gadducci, Angiolo et al. 2000. “Intraperitoneal versus Intravenous Cisplatin in Combination with Intravenous Cyclophosphamide and Epidoxorubicin in Optimally Cytoreduced Advanced Epithelial Ovarian Cancer: A Randomized Trial of the Gruppo Oncologico Nord-Ovest.” Gynecologic Oncology 76(2): 157–62.
Gauthier, Jacques Yves et al. 2008. “The Discovery of Odanacatib (MK-0822), a Selective Inhibitor of Cathepsin K.” Bioorganic and Medicinal Chemistry Letters 18(3): 923–28.
Gourley, Charlie, Joan L. Walker, and Helen J. Mackay. 2016. “Update on Intraperitoneal Chemotherapy for the Treatment of Epithelial Ovarian Cancer.” American Society of Clinical Oncology Educational Book (36): 143–51.
Hauser, Alexander S. et al. 2017. “Trends in GPCR Drug Discovery: New Agents, Targets and Indications.” Nature Reviews Drug Discovery 16(12): 829–42.
Hitchcock, Stephen A. 2012. “Structural Modifications That Alter the P-Glycoprotein Efflux Properties of Compounds.” Journal of Medicinal Chemistry 55(11): 4877–95.
Ho, Teresa T. et al. 2023. “Clinical Impact of the CYP2C19 Gene on Diazepam for the Management of Alcohol Withdrawal Syndrome.” Journal of Personalized Medicine 13(2).
Huang, Guang, Devon Hucek, Tomasz Cierpicki, and Jolanta Grembecka. 2023. “Applications of Oxetanes in Drug Discovery and Medicinal Chemistry.” European Journal of Medicinal Chemistry 261.
Hughes, J. P., S. S. Rees, S. B. Kalindjian, and K. L. Philpott. 2011. “Principles of Early Drug Discovery.” British Journal of Pharmacology 162(6): 1239–49.
Hukkanen, Janne, Peyton Jacob, and Neal L. Benowitz. 2005. “Metabolism and Disposition Kinetics of Nicotine.” Pharmacological Reviews 57(1): 79–115.
Richardson, Samantha, April Bai, Ashutosh A. Kulkarni, and Mehran F. Moghaddam. 2016. “Efficiency in Drug Discovery: Liver S9 Fraction Assay As a Screen for Metabolic Stability.” Drug Metabolism Letters 10(2): 83–90.
Janov, Petra, and Michal iller. 2012. “Phase II Drug Metabolism.” In Topics on Drug Metabolism,.
Jedwabny, Wiktoria, and Edyta Dyguda-Kazimierowicz. 2019. “Revisiting the Halogen Bonding between Phosphodiesterase Type 5 and Its Inhibitors.” Journal of Molecular Modeling 25(2).
Jeffrey, P. 2004. “The Practice of Medicinal Chemistry.” British Journal of Clinical Pharmacology 57(5): 662–662.
Junod, Suzanne. 2008. “FDA and Clinical Drug Trials: A Short History.” Quick Guide to Clinical Trials: 25–55. www.fda.gov.
Kalgutkar, Amit S. 2020. “Designing around Structural Alerts in Drug Discovery.” Journal of Medicinal Chemistry 63(12): 6276–6302.
Kalra, Arjun, and Faiz Tuma. 2018. StatPearls Physiology, Liver. http://www.ncbi.nlm.nih.gov/pubmed/30571059.
Kenny, Peter W. 2019. “The Nature of Ligand Efficiency.” Journal of Cheminformatics 11(1).
Kikuchi, Satoshi et al. 2016. “Prolonged Hypotension Induced by Ingesting a Topical Minoxidil Solution: Analysis of Minoxidil and Its Metabolites.” Acute Medicine & Surgery 3(4): 384–87.
Kim, Jonathan, Rosemary J. Cater, Brendon C. Choy, and Filippo Mancia. 2021. “Structural Insights into Transporter-Mediated Drug Resistance in Infectious Diseases.” Journal of Molecular Biology 433(16).
Kirk, Kenneth. 2006. “Selective Fluorination in Drug Design and Development: An Overview of Biochemical Rationales.” Current Topics in Medicinal Chemistry 6(14): 1447–56.
Kirsch, Philine, Alwin M. Hartman, Anna K.H. Hirsch, and Martin Empting. 2019. “Concepts and Core Principles of Fragment-Based Drug Design.” Molecules 24(23).
Kumari, Shikha, Angelica V. Carmona, Amit K. Tiwari, and Paul C. Trippier. 2020. “Amide Bond Bioisosteres: Strategies, Synthesis, and Successes.” Journal of Medicinal Chemistry 63(21): 12290–358.
Liang, Jun et al. 2016. “Lead Optimization of a pyrazolo[1,5-A]pyrimidin-7(4H)-One Scaffold to Identify Potent, Selective and Orally Bioavailable KDM5 Inhibitors Suitable for in Vivo Biological Studies.” Bioorganic and Medicinal Chemistry Letters 26(16): 4036–41.
Limban, Carmen et al. 2018. “The Use of Structural Alerts to Avoid the Toxicity of Pharmaceuticals.” Toxicology Reports 5: 943–53.
Lipinski, Christopher A. 2004. “Lead- and Drug-like Compounds: The Rule-of-Five Revolution.” Drug Discovery Today: Technologies 1(4): 337–41.
Madden, Judith C., Steven J. Enoch, Alicia Paini, and Mark T.D. Cronin. 2020. “A Review of In Silico Tools as Alternatives to Animal Testing: Principles, Resources and Applications.” ATLA Alternatives to Laboratory Animals 48(4): 146–72.
Magalhães, Pedro R. et al. 2021. “Identification of Pan-Assay INterference compoundS (PAINS) Using an MD-Based Protocol.” In Methods in Molecular Biology, , 263–71.
Mahanta, Nilkamal, D. Miklos Szantai-Kis, E. James Petersson, and Douglas A. Mitchell. 2019. “Biosynthesis and Chemical Applications of Thioamides.” ACS Chemical Biology.
Di Martino, Rita Maria Concetta, Brad D. Maxwell, and Tracey Pirali. 2023. “Deuterium in Drug Discovery: Progress, Opportunities and Challenges.” Nature Reviews Drug Discovery 22(7): 562–84.
Matthews, Holly, James Hanison, and Niroshini Nirmalan. 2016. “‘Omics’-Informed Drug and Biomarker Discovery: Opportunities, Challenges and Future Perspectives.” Proteomes 4(3).
McEneny-King, Alanna, Andrea N. Edginton, and Praveen P.N. Rao. 2015. “Investigating the Binding Interactions of the Anti-Alzheimer’s Drug Donepezil with CYP3A4 and P-Glycoprotein.” Bioorganic and Medicinal Chemistry Letters 25(2): 297–301.
Meanwell, Nicholas A. 2011. “Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design.” Journal of Medicinal Chemistry 54(8): 2529–91.
Meanwell, Nicholas A.. 2014. “The Influence of Bioisosteres in Drug Design: Tactical Applications to Address Developability Problems.” Topics in Medicinal Chemistry 9: 283–382.
Meunier, Bernard, Samuël P. de Visser, and Sason Shaik. 2004. “Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes.” Chemical Reviews 104(9): 3947–80.
Mykhailiuk, Pavel K. 2019. “Saturated Bioisosteres of Benzene: Where to Go Next?” Organic and Biomolecular Chemistry 17(11): 2839–49.
Nikaido, Hiroshi. 2018. “RND Transporters in the Living World.” Research in Microbiology 169(7–8): 363–71.
Oprea, Tudor I., Andrew M. Davis, Simon J. Teague, and Paul D. Leeson. 2001. “Is There a Difference between Leads and Drugs? A Historical Perspective.” Journal of Chemical Information and Computer Sciences 41(3–6): 1308–15.
Orlova, Anna et al. 2019. “Direct Targeting Options for STAT3 and STAT5 in Cancer.” Cancers 11(12): 1930.
Ortwine, Daniel F., and Ignacio Aliagas. 2013. “Physicochemical and DMPK in Silico Models: Facilitating Their Use by Medicinal Chemists.” Molecular Pharmaceutics 10(4): 1153–61.
Pfaff, Annalise R., Justin Beltz, Emily King, and Nuran Ercal. 2019. “Medicinal Thiols: Current Status and New Perspectives.” Mini-Reviews in Medicinal Chemistry 20(6): 513–29.
Pinheiro, Pedro de Sena Murteira, Lucas Silva Franco, and Carlos Alberto Manssour Fraga. 2023. “The Magic Methyl and Its Tricks in Drug Discovery and Development.” Pharmaceuticals 16(8).
Pirali, Tracey, Marta Serafini, Sarah Cargnin, and Armando A. Genazzani. 2019. “Applications of Deuterium in Medicinal Chemistry.” Journal of Medicinal Chemistry 62(11): 5276–97.
Polanski, Jaroslaw, Aleksandra Tkocz, and Urszula Kucia. 2017. “Beware of Ligand Efficiency (LE): Understanding LE Data in Modeling Structure-Activity and Structure-Economy Relationships.” Journal of Cheminformatics 9(1).
Prachayasittikul, Veda, and Virapong Prachayasittikul. 2016. “P-Glycoprotein Transporter in Drug Development.” EXCLI Journal 15: 113–18.
Qiao, Jennifer X. et al. 2008. “Achieving Structural Diversity Using the Perpendicular Conformation of Alpha-Substituted Phenylcyclopropanes to Mimic the Bioactive Conformation of Ortho-Substituted Biphenyl P4 Moieties: Discovery of Novel, Highly Potent Inhibitors of Factor Xa.” Bioorganic and Medicinal Chemistry Letters 18(14): 4118–23.
Ress, David C., Miles Congreve, Christopher W. Murray, and Robin Carr. 2004. “Fragment-Based Lead Discovery.” Nature Reviews Drug Discovery 3(8): 660–72.
Roskoski, Robert. 2016. “Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes.” Pharmacological Research 103: 26–48.
Sneader, W. 2000. “The Discovery of Aspirin: A Reappraisal.” British Medical Journal 321(7276): 1591–94.
Sohlenius-Sternbeck, Anna-Karin et al. 2016. “Optimizing DMPK Properties: Experiences from a Big Pharma DMPK Department.” Current Drug Metabolism 17(3): 253–70.
Subbaiah, Murugaiah A.M., and Nicholas A. Meanwell. 2021. “Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug Design.” Journal of Medicinal Chemistry 64(19): 14046–128.
Topiol, Sid. 2018. “Current and Future Challenges in GPCR Drug Discovery.” In Methods in Molecular Biology, , 1–21.
Tortora, Gerard J, and Bryan Derrickson. 2014. “Tortora Principles of Anatomy and Physiology 14th Edition.” Physiology 86(10): 555.
Umezawa, Koji, and Isao Kii. 2021. “Druggable Transient Pockets in Protein Kinases.” Molecules 26(3).
Vaz, Alfin. 2005. “Multiple Oxidants in Cytochrome P450 Catalyzed Reactions Implications for Drug Metabolism.” Current Drug Metabolism 2(1): 1–16.
Walsh, C. T. et al. 1996. “Bacterial Resistance to Vancomycin: Five Genes and One Missing Hydrogen Bond Tell the Story.” Chemistry and Biology 3(1): 21–28.
Wang, Hui et al. 2019. “Glutathione Conjugation and Protein Adduction Derived from Oxidative Debromination of Benzbromarone in Mice.” Drug Metabolism and Disposition 47(11): 1281–90.
Wermuth, Camille Georges, David Aldous, Pierre Raboisson, and Didier Rognan. 2015. The Practice of Medicinal Chemistry: Fourth Edition The Practice of Medicinal Chemistry: Fourth Edition.
Wiesenfeldt, Mario P. et al. 2023. “General Access to Cubanes as Benzene Bioisosteres.” Nature 618(7965): 513–18.
Wilhelm, Scott et al. 2007. “Erratum: Discovery and Development of Sorafenib: A Multikinase Inhibitor for Treating Cancer.” Nature Reviews Drug Discovery 6(2): 168–168.
Wójcikowski, Jacek, Patrick Maurel, and Władysława A. Daniel. 2006. “Characterization of Human Cytochrome P450 Enzymes Involved in the Metabolism of the Piperidine-Type Phenothiazine Neuroleptic Thioridazine.” Drug Metabolism and Disposition 34(3): 471–76.
Wrobleski, Stephen T. et al. 2019. “Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165.” Journal of Medicinal Chemistry 62(20): 8973–95.
Yue, Qin et al. 2013. “Evaluation of Metabolism and Disposition of GDC-0152 in Rats Using 14C Labeling Strategy at Two Different Positions: A Novel Formation of Hippuric Acid from 4-Phenyl-5-Amino-1,2,3-Thiadiazoles.” Drug Metabolism and Disposition 41(2): 508–17.
Zhang, Ming Qiang, and Barrie Wilkinson. 2007. “Drug Discovery beyond the ‘Rule-of-Five.’” Current Opinion in Biotechnology 18(6): 478–88.
Cover Image
The image used on the cover of “Test tubes for column chromatography” by Bilal Saqib (CC BY-NC-SA)
Appendix
1
Funding Acknowledgements
This project was funded by the Government of Ontario.
The views expressed in this publication are the views of the authors and do not necessarily reflect those of the Government of Ontario.
Elvin D. de Araujo – University of Toronto Mississauga
Elvin D. de Araujo obtained his PhD from the University of Toronto and completed a postdoctoral fellowship focussed on exploring and targeting oncogenic proteins in the context of structural biology and medicinal chemistry. He currently oversees the Centre for Medicinal Chemistry staff and trainees and facilities their entrepreneurial growth in building spin-off companies to their value inflection point. He has co-authored >45 publications in the fields of medicinal chemistry and biochemistry and has received over 20 academic awards/honors.
Timothy B. Wright – University of Toronto Mississauga
Timothy B. Wright obtained a B.Sc. (Honors Specialization in Chemistry) from The University of Western Ontario in 2014. His graduate studies under the supervision of Professor P. Andrew Evans focused on C–C bond formation and asymmetric catalysis. His research was recognized with an NSERC PGS D Fellowship, and in 2019, he received his Ph.D. from Queen’s University. In 2020, he joined the laboratory of Professor Seth B. Herzon as a postdoctoral fellow at Yale University, where worked on total synthesis and biological evaluation of antiproliferative marine metabolites. Timothy is now a Research Associate at the University of Toronto (Mississauga), where his research interests with Professor Patrick T. Gunning (CMC) include the development of covalent therapeutics, chemoproteomics, and bifunctional ligands.
Bilal Saqib – University of Toronto Mississauga
Bilal is a PhD student at the University of Toronto investigating the undruggable proteome for the development of novel cancer therapeutics under the supervision of Dr. Patrick Gunning. With a keen interest in science communication, Bilal employs photography as a medium for scientific dissemination. With this particular resource, he aims to introduce medicinal chemistry and drug discovery to a wider audience.
Jeffrey W. Keillor – University of Ottawa
Jeffrey W. Keillor began his academic career in 1995 in the Chemistry Department at the Université de Montréal. In 2011 he moved to the Department of Chemistry and Biomolecular Sciences of the University of Ottawa, where he held a University Research Chair in Bioorganic Chemistry until 2021. His research program is situated at the interface of chemistry and biochemistry, in the fields of chemical biology and medicinal chemistry. In 2017 he won the Bernard Belleau Award for his work in the field of medicinal chemistry, and in 2020 he was named a Fellow of the Chemical Institute of Canada (FCIC).
Patrick T. Gunning – University of Toronto Mississauga
Patrick T. Gunning, PhD is a Full Professor at the University of Toronto Mississauga and Director and founder of the Centre for Medicinal Chemistry. Dr. Gunning received his BSc (2001) and PhD (2005) from the University of Glasgow before pursuing postdoctoral studies with Professor Andrew D. Hamilton (Yale University). His research group has focussed on the development of small molecule covalent technologies and he has spun out multiple biotech companies including Janpix Ltd., Dalriada Drug Discovery, and Dunad Therapeutics. He has received multiple awards and has been named in Canada’s Top 40 under 40 awardees.