Attributions des images
Figure 2.1 (Figure 1) par Raquel Guillamat-Prats est utilisée sous licence CC-BY 1.
Figure 2.4 (Fig 1) par Osamu Kumano, Kohei Akatsuchi et Jean Amiral est utilisée sous licence CC-BY 4.0.
Figure 3.2 (Fig 2) par Stamatios Lampsas, Maria Xenou, Evangelos Oikonomou, Panteleimon Pantelidis, Antonios Lysandrou, Savvas Sarantos, Athina Goliopoulou, Konstantinos Kalogera, Vasiliki Tsigkou, Athanasios Kalpis, Stavroula A. Paschou, Panagiotis Theofilis, Manolis Vavuranakis, Dimitris Tousoulis et Gerasimos Siasos est utilisée sous licence CC-BY 4.0.
Figure 3.3 (Fig 1) par Hayato Tada, Atsushi Nohara et Masa-aki Kawashiri est utilisée sous licence CC-BY 4.0.
Figure 3.4 (Fig 1) par Dragos Cretoiu, Ruxandra Florentina Ionescu, Robert Mihai Enache, Sanda Maria Cretoiu et Silviu Cristian Voinea est utilisée sous licence CC-BY 4.0.
Figure 3.5 (Fig 1) par Simona Pisanti, Erika Rimondi, Elena Pozza, Elisabetta Melloni, Enrico Zauli, Maurizio Bifulco, Rosanna Martinelli et Annalisa Marcuzzi est utilisée sous licence CC-BY 4.0.
Figure 3.8 (Fig 12) par Marcel Hrubša, Tomáš Siatka, Iveta Nejmanová, Marie Vopršalová, Lenka Kujovská Krčmová, Kateřina Matoušová, Lenka Javorská, Kateřina Macáková, Laura Mercolini, Fernando Remião, Marek Máťuš, Přemysl Mladěnka a été modifiée (recadrée) et est utilisée sous licence CC-BY 4.0.
Figure 3.9 (Fig 1) par Jun Zeng, Wenjing Liu, Bing Liang, Lingyu Shi, Shanbo Yang, Jingsen Meng, Jing Chang, Xiaokun Hu, Renshuai Zhang, et Dongming Xing a été modifiée (recadrée) et est utilisée sous licence CC-BY 4.0.
Figure 3.11 (Fig 1) par Simona Scheggi, Graziano Pinna, Giulia Braccagni, Maria Graziella De Montis et Carla Gambarana est utilisée sous licence CC-BY 4.0.
Figure 4.1 (Fig 5) par Katharina Schreck et Matthias F. Melzig est utilisée sous licence CC-BY 4.0.
Figure 4.2 (Fig 3) par Joanna Michałowska, Ewa Miller-Kasprzak et Paweł Bogdański est utilisée sous licence CC-BY 4.0.
Figure 4.3 (Fig 1) par Ji-Hye Lee et Jaemin Lee est utilisée sous licence CC-BY 4.0.
Figure 4.4 (Fig 1) par Selvaraj Jayaraman, Anitha Roy, Srinivasan Vengadassalapathy, Ramya Sekar, Vishnu Priya Veeraraghavan, Ponnulakshmi Rajagopal, Gayathri Rengasamy, Raktim Mukherjee, Durairaj Sekar et Reji Manjunathan est utilisée sous licence CC-BY 4.0.
Figure 4.5 (Fig 1) par Dariusz Szukiewicz est utilisée sous licence CC-BY 4.0.
Figure 4.6 (Fig 1) par Beatrice Rosetti et Silvia Marchesan est utilisée sous licence CC-BY 4.0.
Figure 4.6 (Fig 2) par Harish Vashisth a été modifiée (recadrée) et utilisée sous licence CC-BY 4.0.
Figure 4.7 (Fig 4) par Takuo Ogihara, Kenta Mizoi et Akiko Ishii-Watabe est utilisée sous licence CC-BY 4.
Figure 4.8 (Fig 5) par Takuo Ogihara, Kenta Mizoi et Akiko Ishii-Watabe est utilisée sous licenceCC-BY 4.
Figure 4.10 (Fig 1) par Ichiro Nojima et Jun Wada est utilisée sous licence CC-BY 4.0.
Figure 4.14 (Fig 2) par Wenwei Wan, Qikai Qin, Linshan Xie, Hanqing Zhang, Fan Wu, Raymond C. Stevens et Yan Liu a été modifiée (recadrée) et est utilisée sous licences CC-BY 4.0.
Figure 4.16 (Fig 1) par Daria M. Keller, Natasha Ahmed, Hamza Tariq, Malsha Walgamage, Thilini Walgamage, Azad Mohammed, Jadzia Tin-Tsen Chou, Marta Kałużna-Oleksy, Maciej Lesiak et Ewa Straburzyńska-Migaj est utilisée sous licence CC-BY 4.0.
Figure 4.19 (Fig 1) par Tapan Behl, Piyush Madaan, Aayush Sehgal, Sukhbir Singh, Neelam Sharma, Saurabh Bhatia, Ahmed Al-Harrasi, Sridevi Chigurupati, Ibrahim Alrashdi et Simona Gabriela Bungau est utilisée sous licence CC-BY 4.0.
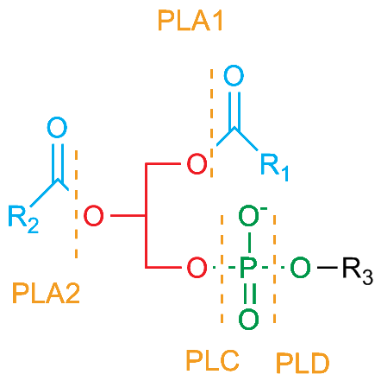
Figure 5.1 (Phospholipases2) par Roadnottaken est utilisée sous licence CC BY-SA 3.0.
Figure 5.3 (Fig 1) par Cándido Ortiz-Placín, Alba Castillejo-Rufo, Matías Estarás et Antonio González est utilisée sous licence CC-BY 4.0.
Figure 5.4 (Fig 1) par Inger L. Meek, Mart A.F.J. Van de Laar et Harald E. Vonkeman est utilisée sous licence CC-BY 3.0.
Figure 6.1 (Fig 1) par Dariusz Szukiewicz est utilisée sous licence CC-BY 4.0.
Références bibliographiques
Adcock, D. M., et R. Gosselin. (2015). « Direct oral anticoagulants (DOACs) in the laboratory: 2015 review ». Thrombosis Research, 136(1), 7-12.
Ayoub, Samir S. (2021). « Paracetamol (Acetaminophen): A familiar drug with an unexplained mechanism of action. » Temperature, 8(4), 351-71.
Bailey, Clifford J. (2017). « Metformin: historical overview. » Diabetologia, 60(9), 1566-76.
Burnett, Allison E. et coll. (2016). « Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment ». Journal of Thrombosis and Thrombolysis, 41(1), 206-32.
Cavallari, Larisa H. et Minoli A. Perera. (2012). « The future of warfarin pharmacogenetics in under-represented minority groups ». Future Cardiology, 8(4), 563-76.
Cretoiu, Dragos et coll. (2021). « Gut microbiome, functional food, atherosclerosis, and vascular calcifications—is there a missing link? » Microorganisms, 9(9).
Druker, Brian J. (2002). « STI571 (GleevecTM) as a paradigm for cancer therapy ». Trends in Molecular Medicine, 8(4 SUPPL.).
Durrington, Paul et Handrean Soran. (2014). « Hyperlipidemia ». Metabolism of Human Diseases: Organ Physiology and Pathophysiology, 295-302.
Endo, A. (1992). « The discovery and development of HMG-CoA reductase inhibitors ». Journal of Lipid Research, 33(11), 1569-82.
Farnier, Michel et Jean Davignon. (1998). « Current and future treatment of hyperlipidemia: the role of statins ». American Journal of Cardiology, 3J-10J.
Gage, Brian F., Stephan D. Fihn et Richard H. White. (2000). « Management and dosing of warfarin therapy ». American Journal of Medicine, 109(6), 481-88.
Greinacher, Andreas. (2009). « Heparin-induced thrombocytopenia ». Journal of Thrombosis and Haemostasis, 7(SUPPL. 1), 9-12.
Grinstein, Jonathan D. (2023). « The immaculate conception of Gleevec, as told by Brian Druker ». Genetic Engineering and Biotechnology News, 5(1), 325-28.
Gröber, U., J. Reichrath, M. F. Holick et K. Kisters. (2014). « Vitamine K: an old vitamin in a new perspective ». Dermato-Endocrinology, 6(1).
Guillamat-Prats, Raquel. (2022). « Role of mesenchymal stem/stromal cells in coagulation ». International Journal of Molecular Sciences, 23(18).
Hemker, H. C. (2016). « A century of heparin: past, present and future ». Journal of Thrombosis and Haemostasis, 14(12), 2329-38.
Hrubša, Marcel et coll. (2022). « Biological properties of vitamins of the B-Complex, part 1: vitamins B1, B2, B3, and B5 ». Nutrients, 14(3).
Ivanova, Donika et coll. (2018). « Vitamine K: redox-modulation, prevention of mitochondrial dysfunction and anticancer effect ». Redox Biology, 16, 352-58.
Kelton, John G. et Theodore E. Warkentin. (2008). « Heparin-induced thrombocytopenia: a historical perspective ». Blood, 112(7), 2607-16.
Khavandi, Kaivan et coll. (2014). « Interrupting the natural history of diabetes mellitus: lifestyle, pharmacological and surgical strategies targeting disease progression ». Current Vascular Pharmacology, 12(1), 155-67.
Kriaa, Aicha et coll. (2022). « Bile acids: key players in inflammatory bowel diseases? » Cells, 11(5).
Kumano, Osamu, Kohei Akatsuchi et Jean Amiral. (2021). « Updates on anticoagulation and laboratory tools for therapy monitoring of heparin, vitamin K antagonists and direct oral anticoagulants ». Biomedicines, 9(3).
L, Dean. (2018). « Warfarin therapy and VKORC1 and CYP genotype ». Medical Genetics Summaries (Md), 1-9. http://europepmc.org/books/NBK84174%0Ahttps://europepmc.org/article/NBK/nbk84174.
Lampsas, Stamatios et coll. (2023). « Lipoprotein(a) in atherosclerotic diseases: from pathophysiology to diagnosis and treatment ». Molecules, 28(3).
Lee, Catherine J. et Jack E. Ansell. (2011). « Direct thrombin inhibitors ». British Journal of Clinical Pharmacology, 72(4), 581-92.
Lee, Ji Hye et Jaemin Lee. (2022). « Endoplasmic reticulum (ER) stress and its role in pancreatic β‐cell dysfunction and senescence in type 2 diabetes ». International Journal of Molecular Sciences, 23(9).
Lee, William M. (2020). « Acetaminophen toxicity: a history of serendipity and unintended consequences ». Clinical Liver Disease, 16(S1), 34-44.
Lin, Yen Lin, Yilin Meng, Wei Jiang et Benoît Roux. (2013). « Explaining why Gleevec is a specific and potent inhibitor of Abl kinase ». Proceedings of the National Academy of Sciences of the United States of America, 110(5), 1664-69.
Lutsey, Pamela L. et Neil A. Zakai. (2023). « Epidemiology and prevention of venous thromboembolism ». Nature Reviews Cardiology, 20(4), 248-62.
Martinelli, Ida, Paolo Bucciarelli et Pier Mannuccio Mannucci. (2010). « Thrombotic risk factors: basic pathophysiology ». Critical Care Medicine, 38(SUPPL. 2).
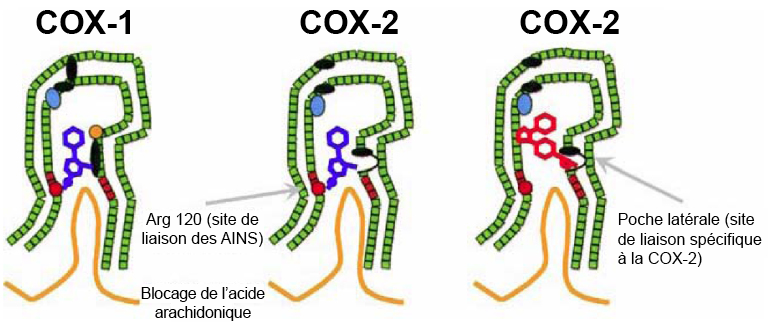
Meek, Inger L., Mart A.F.J. van de Laar et Harald E. Vonkeman. (2010). « Non-steroidal anti-inflammatory drugs: an overview of cardiovascular risks ». Pharmaceuticals, 3(7), 2146-62.
Michałowska, Joanna, Ewa Miller-Kasprzak et Paweł Bogdański. (2021). « Incretin hormones in obesity and related cardiometabolic disorders: the clinical perspective ». Nutrients, 13(2), 1-32.
Mueckler, Mike et Bernard Thorens. (2013). « The SLC2 (GLUT) family of membrane transporters ». Molecular Aspects of Medicine, 34(2–3), 121-38.
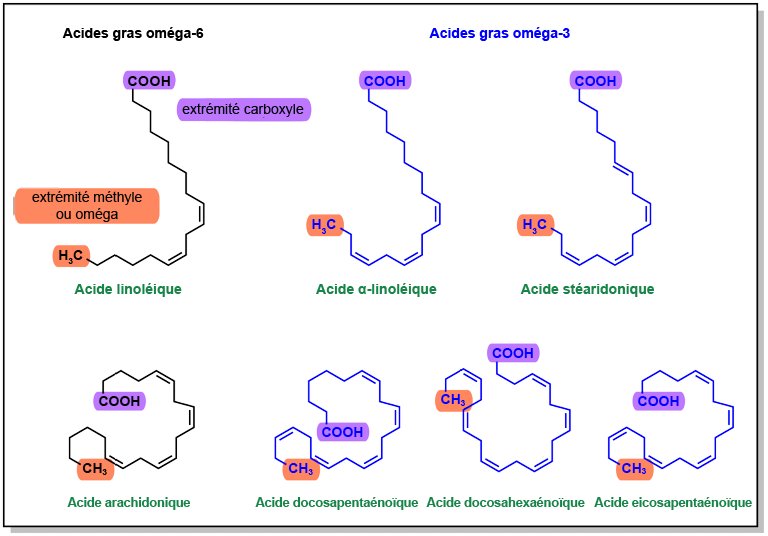
Nakamura, M T et T Y Nara. (2004). « Structure, function, and dietary regulation of delta6, delta5, and delta9 Desaturases ». Annu Rev Nutr, 24, 345-76.
Nawarskas, James J. (2005). « HMG-CoA reductase inhibitors and coenzyme Q10 ». Cardiology in Review, 13(2), 76-79.
Nojima, Ichiro et Jun Wada. (2023). « Metformin and its immune-mediated effects in various diseases ». International Journal of Molecular Sciences, 24(1).
Ogihara, Takuo, Kenta Mizoi et Akiko Ishii-Watabe. (2023). « Pharmacokinetics of biopharmaceuticals: their critical role in molecular design ». Biomedicines, 11(5).
Ortel, Thomas L. et coll. (2020). « American Society of Hematology 2020 guidelines for management of venous thromboembolism: treatment of deep vein thrombosis and pulmonary embolism ». Blood Advances, 4(19), 4693-4738.
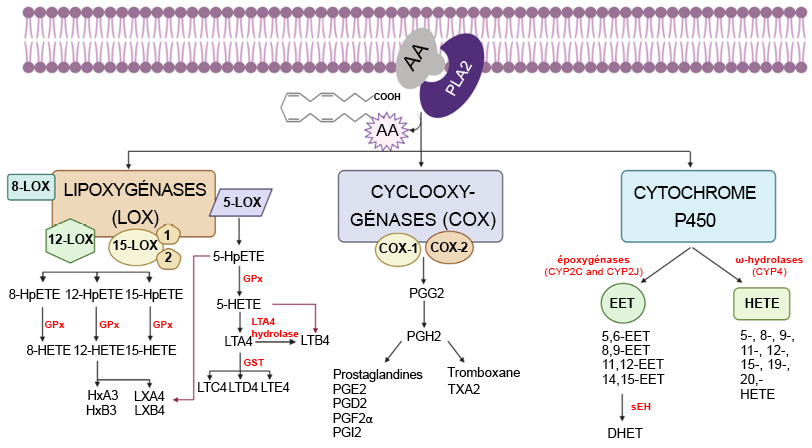
Ortiz-Placín, Cándido, Alba Castillejo-Rufo, Matías Estarás et Antonio González. (2023). « Membrane lipid derivatives: roles of arachidonic acid and its metabolites in pancreatic physiology and pathophysiology ». Molecules, 28(11).
Phan, Binh An P., Thomas D. Dayspring et Peter P. Toth. (2012). « Ezetimibe therapy: mechanism of action and clinical update ». Vascular Health and Risk Management, 8(1), 415-27.
Pisanti, Simona et coll. (2022). « Prenylation defects and oxidative stress trigger the main consequences of neuroinflammation linked to mevalonate pathway deregulation ». International Journal of Environmental Research and Public Health, 19(15).
Porter, William R. (2010). « Warfarin: history, tautomerism and activity ». Journal of Computer-Aided Molecular Design, 24(6-7), 553-73.
Rieg, Timo et Volker Vallon. (2018). « Development of SGLT1 and SGLT2 inhibitors ». Diabetologia, 61(10), 2079-86.
Rosetti, Beatrice et Silvia Marchesan. (2023). « Peptide inhibitors of insulin fibrillation: current and future challenges ». International Journal of Molecular Sciences, 24(2).
Scheggi, Simona et coll. (2022). « PPARα signaling: a candidate target in psychiatric disorder management ». BioMolecules, 12(5).
Schreck, Katharina et Matthias F. Melzig. (2018). « Intestinal saturated long-chain fatty acid, glucose and fructose transporters and their inhibition by natural plant extracts in caco-2 cells ». Molecules, 23(10).
Smith, Francine G., Andrew W. Wade, Megan L. Lewis et Wei Qi. (2012). « Cyclooxygenase (COX) inhibitors and the newborn kidney ». Pharmaceuticals, 5(11), 1160-76.
Staels, Bart et coll. (1998). « Mechanism of action of fibrates on lipid and lipoprotein metabolism ». Circulation, 98(19), 2088-93.
Szukiewicz, Dariusz. (2023). « Molecular mechanisms for the vicious cycle between insulin resistance and the inflammatory response in obesity ». International Journal of Molecular Sciences, 24(12).
Tada, Hayato, Atsushi Nohara et Masa Aki Kawashiri. (2018). « Serum triglycerides and atherosclerotic cardiovascular disease: insights from clinical and genetic studies ». Nutrients, 10(11).
Tobert, Jonathan A. (2003). « Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors ». Nature Reviews Drug Discovery, 2(7), 517-26.
Turunen, Mikael, Jerker Olsson et Gustav Dallner. (2004). « Metabolism and function of coenzyme Q ». Biochimica et Biophysica Acta – Biomembranes, 1660(1-2), 171-99.
Vashisth, Harish. (2015). « Theoretical and computational studies of peptides and receptors of the insulin family ». Membranes, 5(1), 48-83.
Vaz, JuliusA et Ashis Patnaik. (2012). « Diabetes Mellitus: exploring the challenges in the drug development process ». Perspectives in Clinical Research, 3(3), 109.
Wan, Wenwei et coll. (2023). « GLP-1R signaling and functional molecules in incretin therapy ». Molecules, 28(2).
Wardrop, Douglas et David Keeling. (2008). « The story of the discovery of heparin and warfarin ». British Journal of Haematology, 141(6), 757-63.
Williamson, Mike P. (2023). « Protein Binding: a fuzzy concept ». Life, 13(4).
Zeng, Jun et coll. (2022). « Inhibitory effect of isoliquiritigenin in niemann-pick C1-like 1-mediated cholesterol uptake ». Molecules, 27(21).
Zhao, Yanfang et coll. (2015). « Design, synthesis and structure – activity relationship of oxazolidinone derivatives containing novel S4 ligand as FXa inhibitors ». European Journal of Medicinal Chemistry, 96, 369-80.
Image de la page couverture
L’image utilisée sur la couverture est « Dispensing cell culture media » par Bilal Saqib (CC BY-NC-SA)

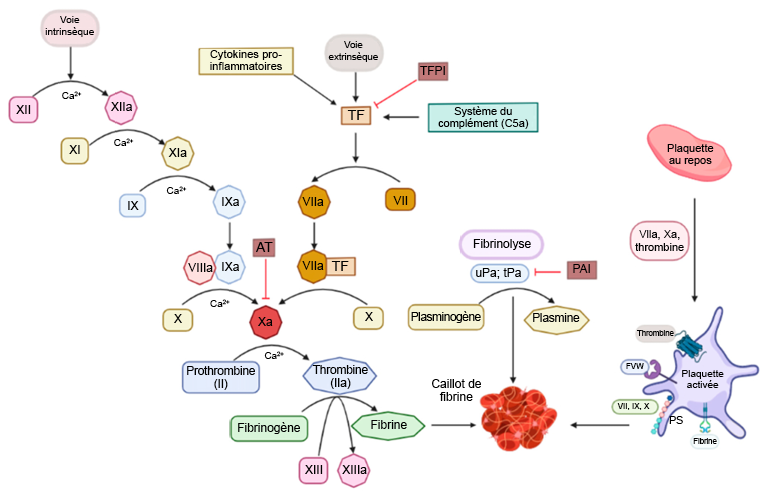
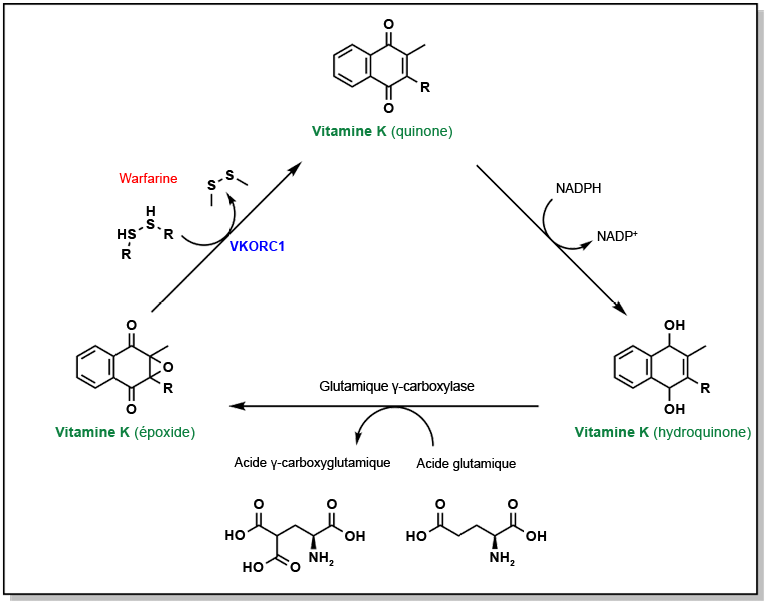
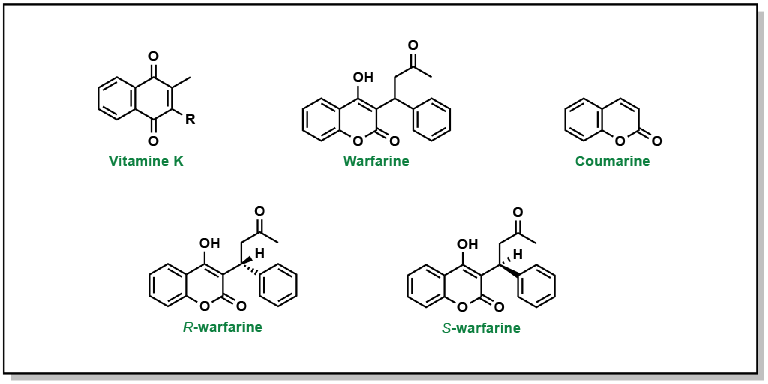
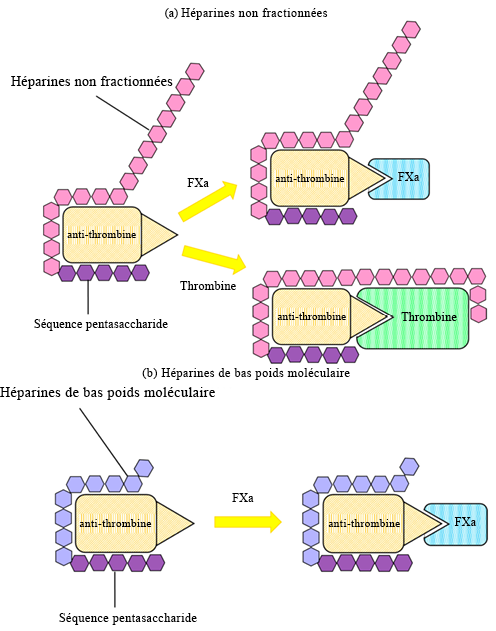
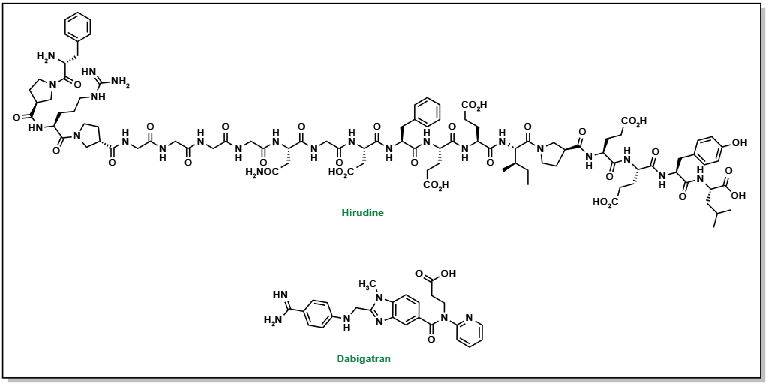
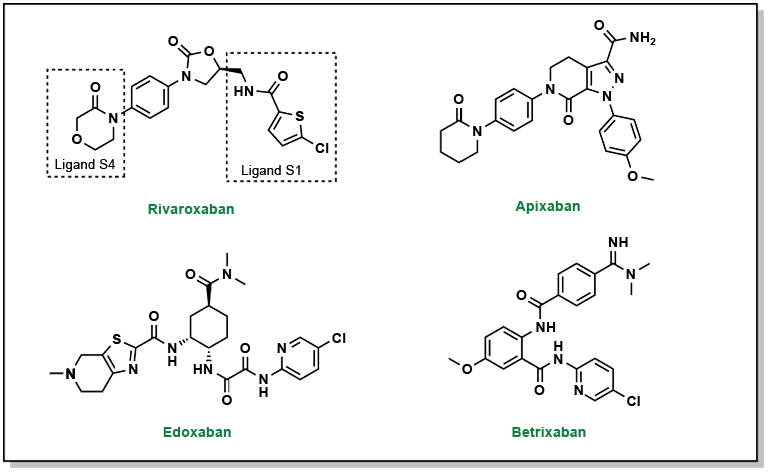
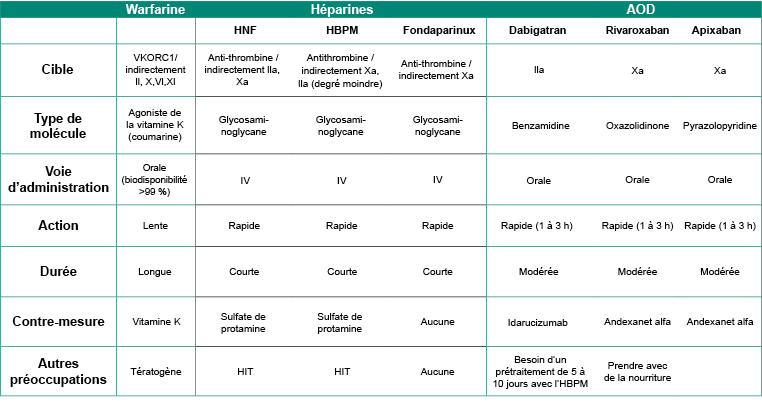
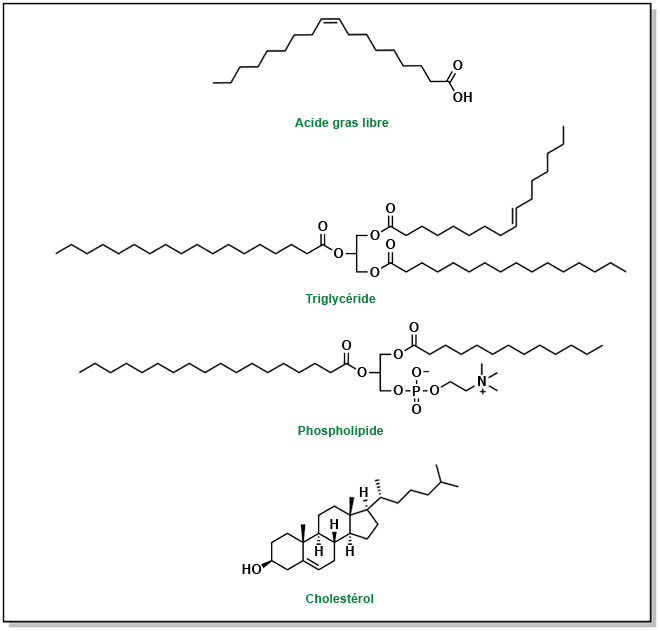
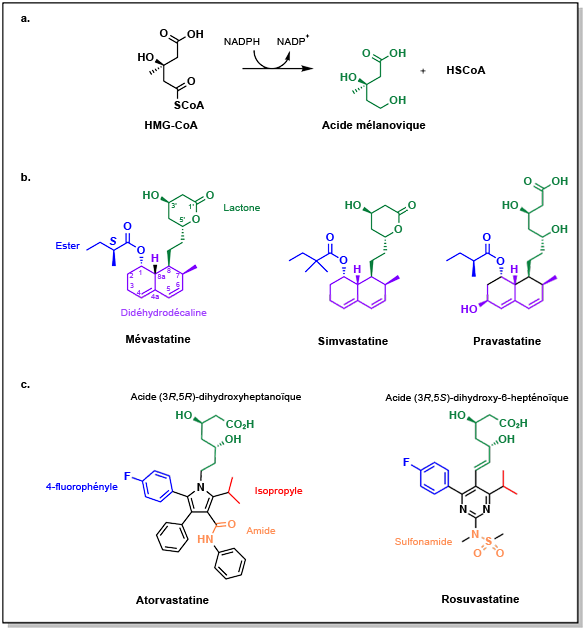
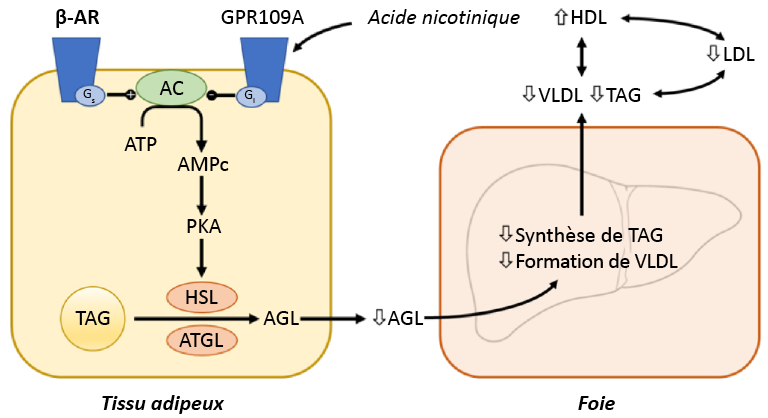
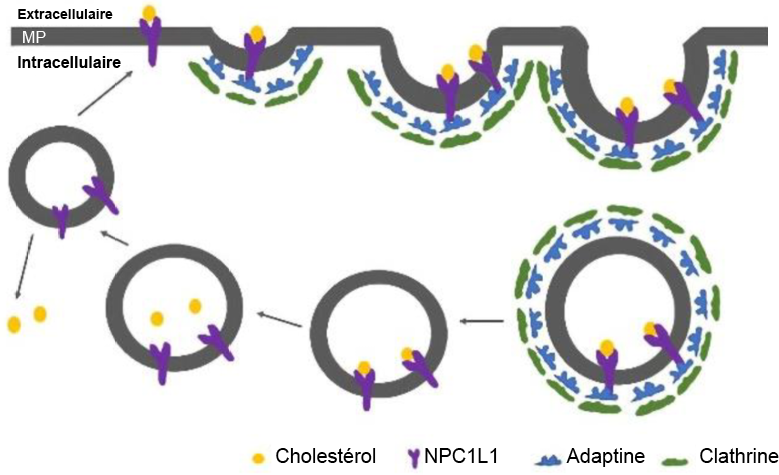
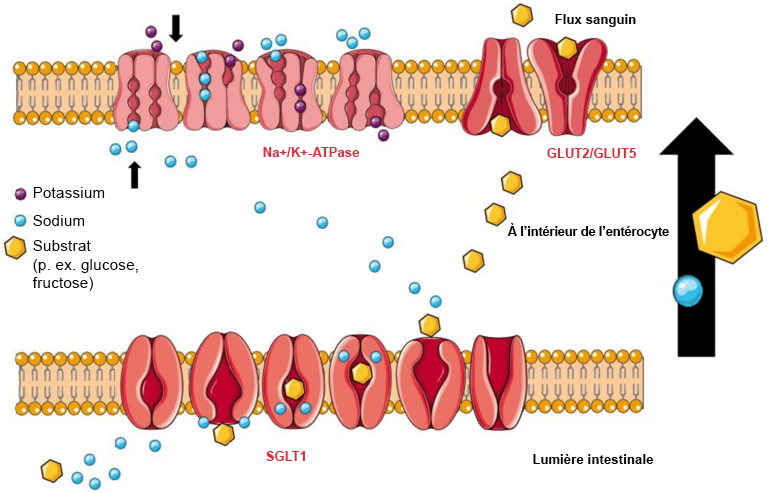
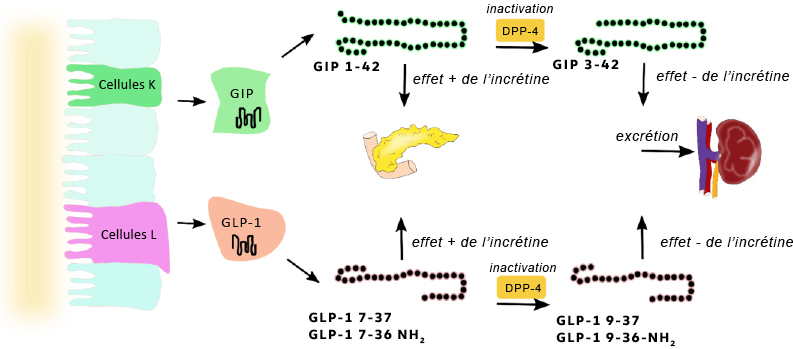
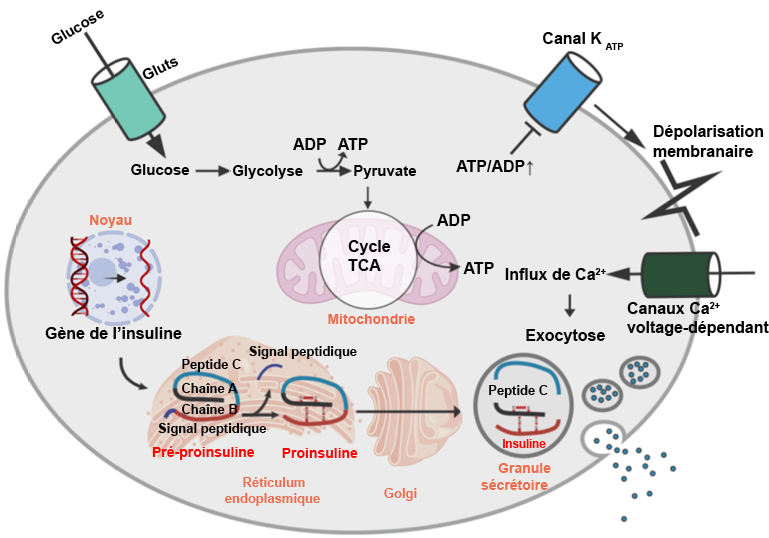
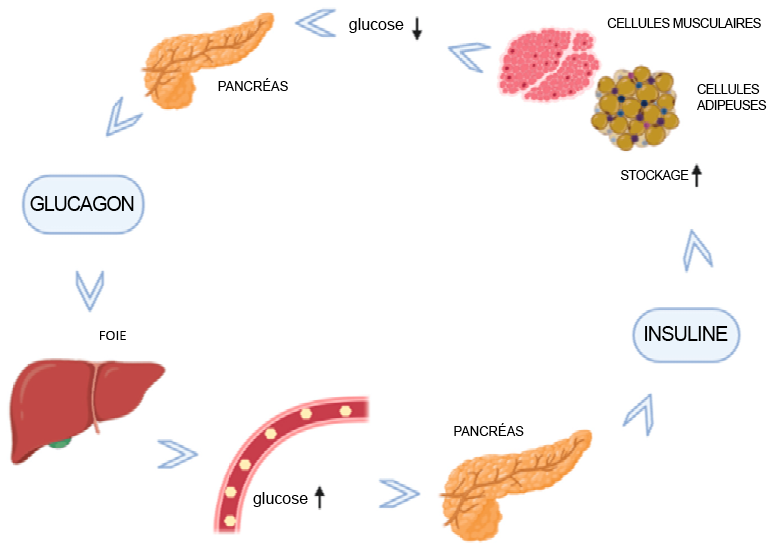
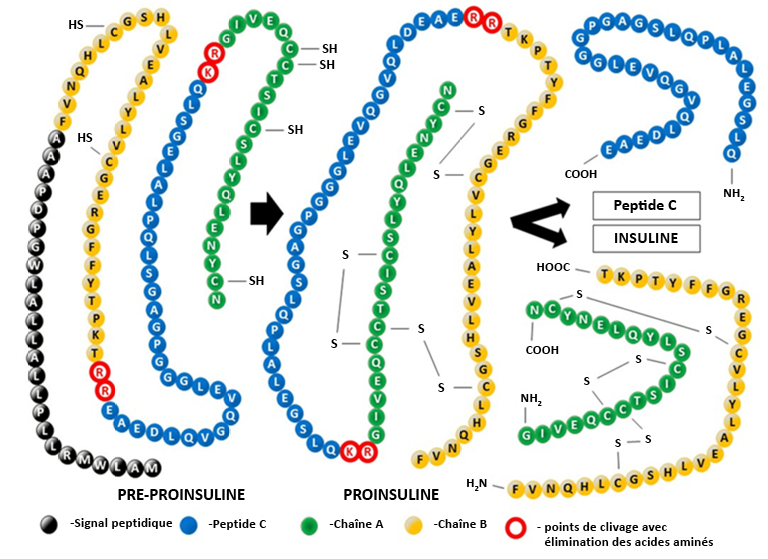
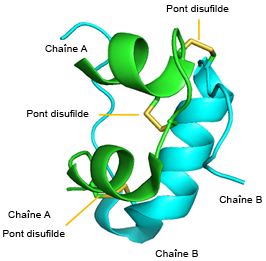
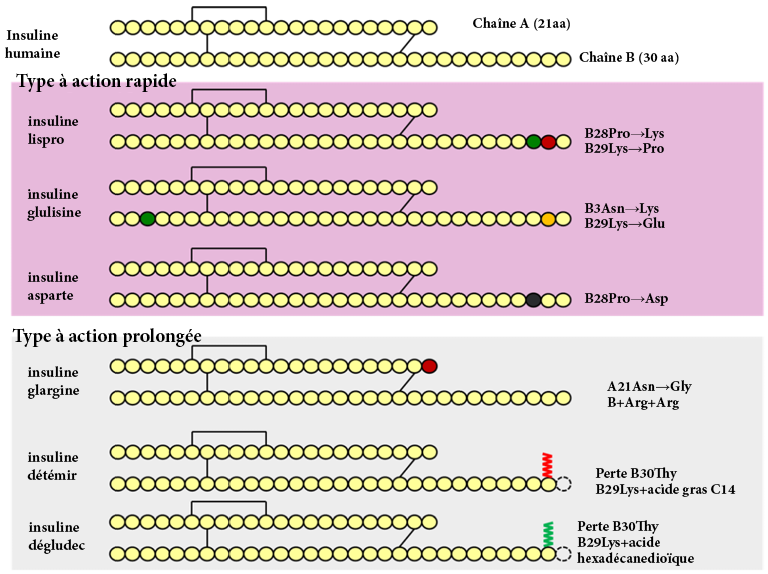
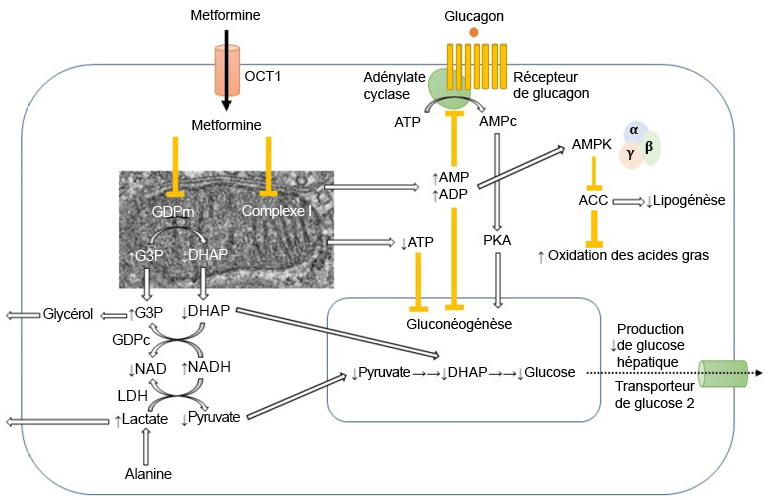 FIGURE 4.10 Différents effets de la metformine dans la cellule. Source de l’image : (
FIGURE 4.10 Différents effets de la metformine dans la cellule. Source de l’image : (

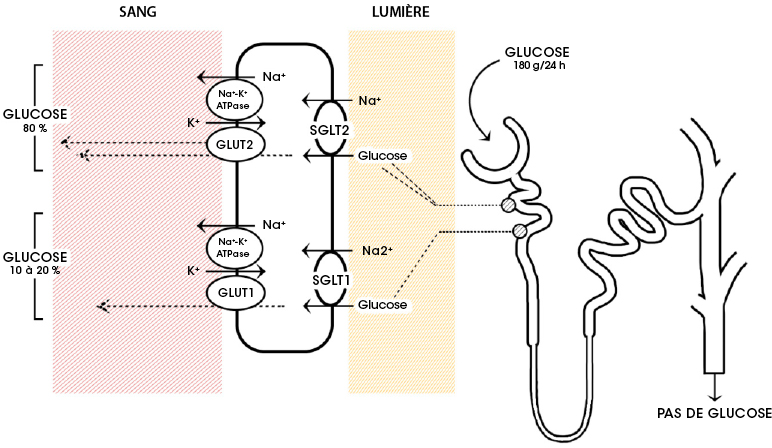 FIGURE 4.16 Réabsorption du glucose par le néphron du rein. Source de l’image : (
FIGURE 4.16 Réabsorption du glucose par le néphron du rein. Source de l’image : (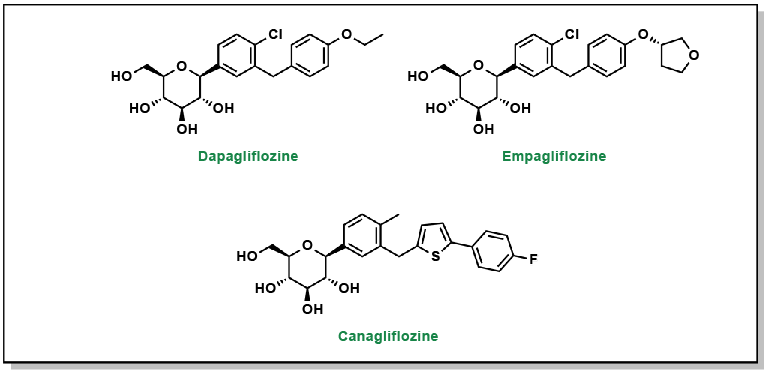
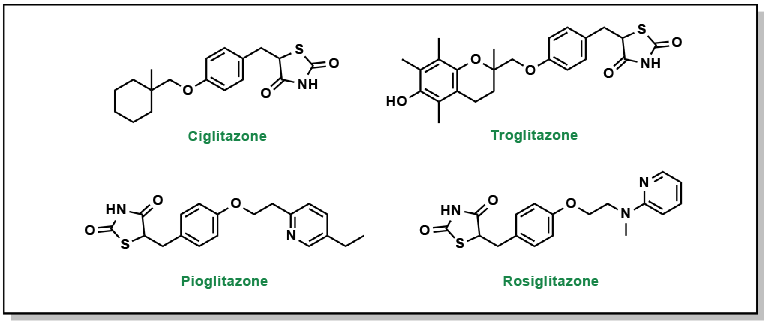 FIGURE 4.18 Structures des thiazolidinediones.
FIGURE 4.18 Structures des thiazolidinediones.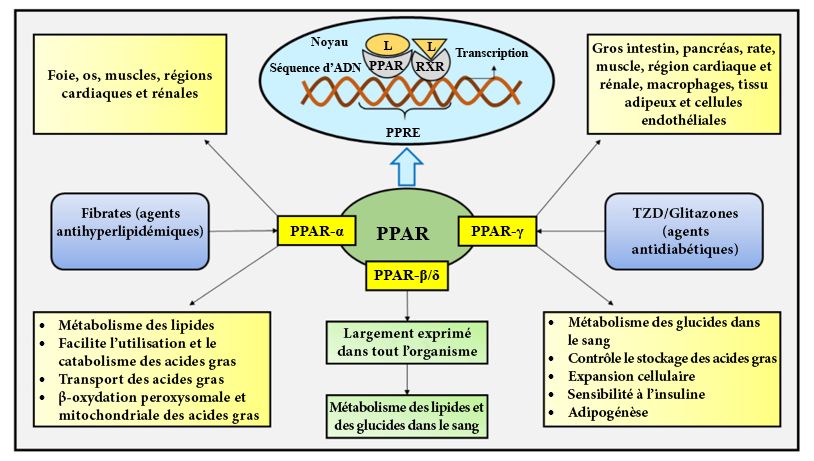
{kind=link}