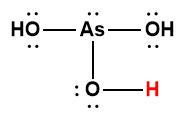
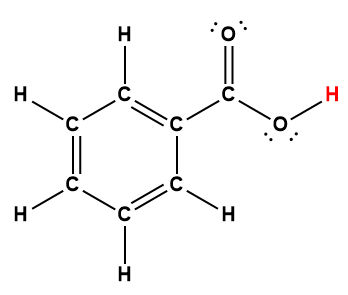
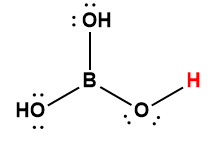
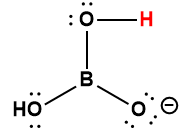
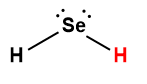
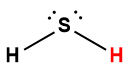
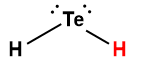
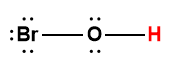
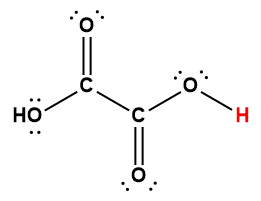
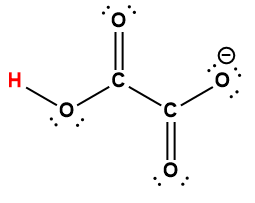
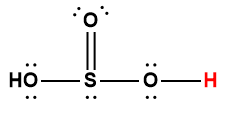
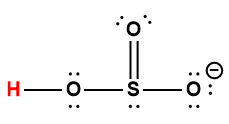
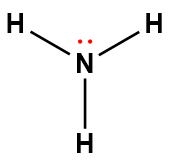
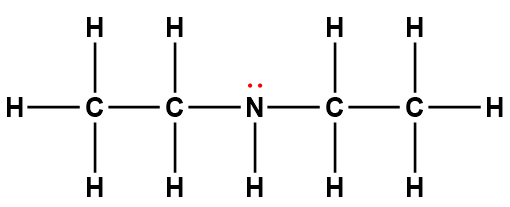
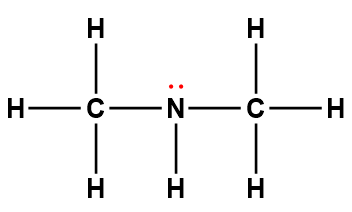
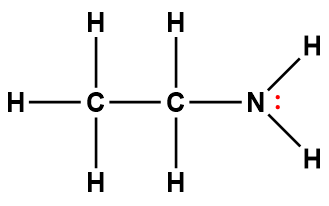
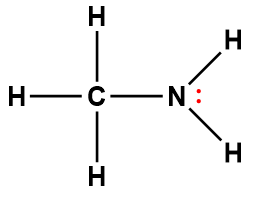
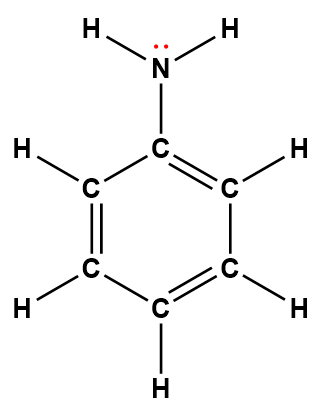
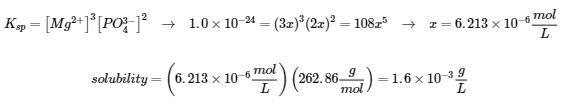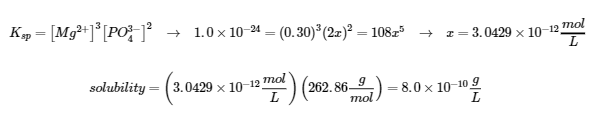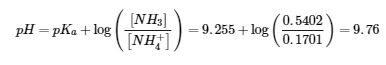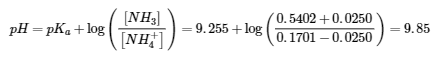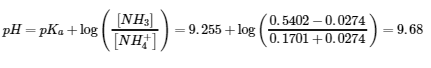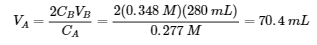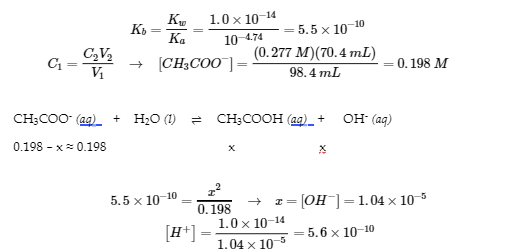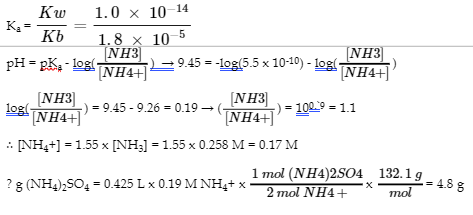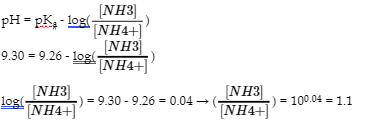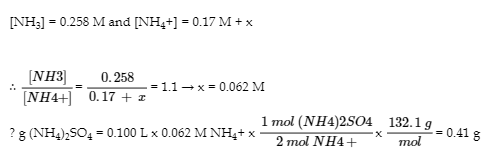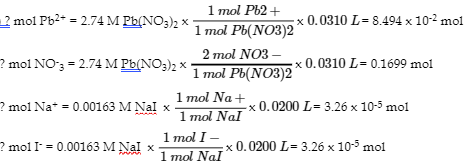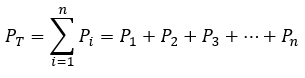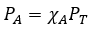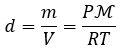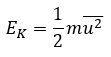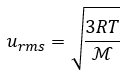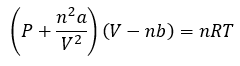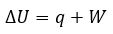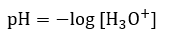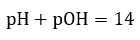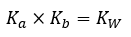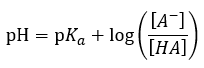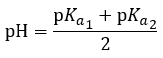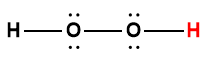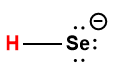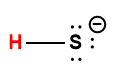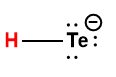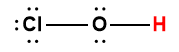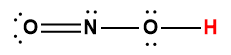Page de Couverture
Les illustrateurs de la page de couverture étaient Sarah Nersesian et Anupreet Kharbanda de Designs that Cell.
Rencontrer les Auteurs
Les illustrateurs des images d’auteur étaient Sarah Nersesian et Anupreet Kharbanda de Designs that Cell.
1 – Stœchiométrie
1.1 – La mole
Ce chapitre contient du matériel et des exercices tirés de la section 3.1 “Formula Mass and the Mole Concept” et ses exercices, respectivement, de la ressource de manuel ouvert Chemistry 2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
Paragraphes 1 à 7,
Exemples 1.1.1, 1.1.2, 1.1.3, 1.1.4, 1.1.5 et 1.1.6,
“Vérifiez votre apprentissage” 1.1.1, 1.1.2, 1.1.3, 1.1.4, 1.1.5 et 1.1.6, et
Tableau 1.1.1.
Ce chapitre contient également des informations tirées de la section 1.5 “Densité et composition en pourcentage” : Leur utilisation dans la résolution de problèmes” de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0, y compris :
Le paragraphe 10, et
Exemple 1.1.7.
Ce chapitre contient des éléments tirés du point 3.2 – Détermination des formules empiriques et moléculaires, y compris les paragraphes 8 et 9.
Ce chapitre contient des éléments tirés de la section 3.1 “Formula Mass and the Mole Concept” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licence CC BY 4.0, y compris la fin de la section 1.1 questions et ses réponses.
Ce chapitre contient des documents originaux de Jessica Thomas, y compris les documents entre parenthèses de l’exemple 1.1.1 et les réponses aux questions 1 et 7 de la fin de la section 1.1.
Ce chapitre contient des documents originaux de Leanne Trepanier et Nathan Biniam, y compris les réponses à la fin de la section 1.1, questions 5 et 8.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des figures, des exemples et des tableaux.
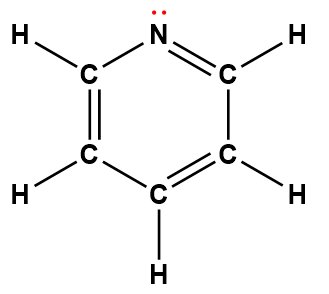
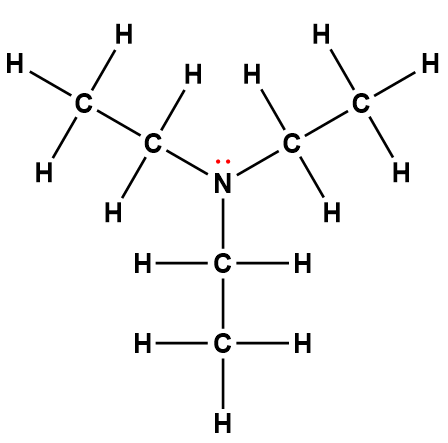
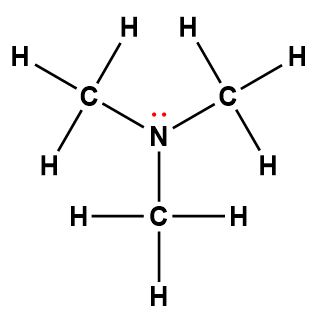
Ce chapitre contient les figures 1.1.1, 1.1.2, 1.1.3 et 1.1.4 tirées de la section 3.1 – Formule de masse et concept de mole.
1.2 – Détermination des formules chimiques
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 2.3 “Structure atomique et symbolisme“.
La boîte “Au cas où vous seriez intéressé … Spectrométrie de masse
Section 2.4 “Formules chimiques” et ses exercices,
Paragraphes 2 à 6 et 11,
Section 3.2 “Déterminer les formules empiriques et moléculaires” et ses exercices,
Paragraphes 12-16 et 18-23,
Exemples 1.2.1, 1.2.2 et 1.2.4,
“Vérifiez votre apprentissage” 1.2.1 et 1.2.2,
Section 4.1 “Rédaction et équilibrage des équations chimiques” et ses exercices,
Paragraphes 24-33,
L’encadré “Équations chimiques équilibrées – pratique supplémentaire”,
Section 4.5 “Analyse chimique quantitative“.
Paragraphe 17,
Exemples 1.2.3 et 1.2.5,
“Vérifiez votre apprentissage” 1.2.3 et 1.2.7,
tous utilisés sous une licenceCC BY 4.0.
Ce chapitre contient également un exercice tiré de la section 1.3 “Introduction to Combustion Analysis” du manuel ouvert Physical Methods in Chemistry and Nano Science (par Raja et Barron) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY 4.0.
Ce chapitre contient également un exemple et des exercices tirés des exercices de la section 3 “Stoechiométrie” de la carte de textes Libretextes pour la chimie : The Central Science (par Brown, LeMay, Busten, Murphy et Woodward) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous licenceCC BY-NC-SA 4.0.
Ce chapitre contient l’exemple 1.2.4 tiré de la section 3.2 – Détermination des formules empiriques et moléculaires.
Ce chapitre contient la fin de la section 1.2, les questions 1 et 2 et leurs réponses tirées de la section 2.4 “Formules chimiques” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient la fin de la section 1.2 questions 3-6 et ses réponses tirées de la section 3.2 “Determining Empiricaland MolecularFormulas” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, and Robinson, PhD, utilisé sous une licenceCC BY 4.0.
Ce chapitre contient la fin de la section 1.2, les questions 7 et 8, et ses réponses tirées des exercices 3. E “Stoechiométrie(Exercices)” de la carte de texte Libretexts pour la chimie : The Central Science (par Brown, LeMay, Busten, Murphy et Woodward) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous licenceCC BY-NC-SA 4.0.
Ce chapitre contient la fin de la section 1.2 question 9-11 et ses réponses tirées de la section 4.1 “Writingand Balancing Chemical Equations” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, and Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient du matériel original écrit par le Dr. Brandi West, y compris le paragraphe sous la réponse de “Vérifiez votre apprentissage” 1.2.6.
Ce chapitre comprend le matériel original écrit par Mahdi Zeghal, y compris les paragraphes 1 et 7-9, les parenthèses de la deuxième phrase du paragraphe 3, les parenthèses de la première phrase du paragraphe 4, les parenthèses de la deuxième phrase du paragraphe 6, les parenthèses de la quatrième phrase du paragraphe 11, les parenthèses de la première phrase du paragraphe 17, la case “problèmes d’analyse de combustion – hypothèse sous-jacente”, le troisième paragraphe sous la solution de “Vérifiez votre apprentissage” 1.2.4 et la première phrase de la case “Équations chimiques équilibrées – pratique supplémentaire”.
Ce chapitre contient des documents originaux de Geneviève O’Keefe, notamment la quatrième phrase du paragraphe 6 et le paragraphe 10.
Ce chapitre contient des documents originaux de Leanne Trepanier et Nathan Biniam, y compris les réponses à la fin de la section 1.2, questions 5 et 11.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des exemples.
Ce chapitre contient la figure 1.2.9 tirée de “Structure atomique et symbolisme“.
Ce chapitre contient les figures 1.2.1, 1.2.2, 1.2.3 et 1.2.4 tirées de “Chemical Formulas“.
Ce chapitre contient les figures 1.2.5, 1.2.6 et 1.2.7 tirées de “Determining Empiricaland MolecularFormulas“.
Ce chapitre contient les figures 1.2.11 et 1.2.12 tirées de “Writingand Balancing Chemical Equations“.
Ce chapitre contient la figure 1.2.8 tirée de “Analyse chimique quantitative“.
1.3 – Stœchiométrie des réactions
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 4.3 “Stœchiométrie des réactions” et ses exercices,
Paragraphes 1 à 8,
Exemples 1.3.1, 1.3.2, 1.3.3, 1.3.4,
“Vérifiez votre apprentissage” 1.3.4,
Fin du chapitre 1.3 questions 1-8, et
Section 4.4 “Rendements de réaction” et ses exercices,
Paragraphes 9 à 15,
Exemples 1.3.5, et 1.3.6,
“Vérifiez votre apprentissage” 1.3.5 (a), et
Fin du chapitre 1.3 questions 9-12,
tous deux utilisés sous une licenceCC BY 4.0.
Ce chapitre contient le contenu original créé par Jessica Thomas, y compris l’exemple 1.3.5 question et réponse pour b, et la question et réponse pour l’exemple 1.3.6.
Ce chapitre contient les réponses originales aux questions 1, 2, 5, 6, 8, 9, 10 et 12 créées par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient le contenu original de Geneviève O’Keefe, y compris la numérotation de certains chiffres et la réponse à la question 5 à la fin de cette section.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient les figures 1.3.1 et 1.3.2 tirées de la section 4.3 “Stœchiométrie des réactions“.
Ce chapitre contient les figures 1.3.3 et 1.3.4 tirées de la section 4.4 “Rendements de réaction“.
1.4 – Stœchiométrie des solutions
Ce chapitre contient du matériel et des exercices tirés de la section 3.3 “Molarité” et de ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0, y compris :
Paragraphes 1-5 et 19-22,
Exemples 1.4.1, 1.4.2, 1.4.3, 1.4.4, 1.4.5, 1.4.11, 1.4.12, 1.4.13, et
“Vérifiez votre apprentissage” 1.4.1, 1.4.2, 1.4.3, 1.4.4, 1.4.5, 1.4.11, 1.4.12, et 1.4.13.
Ce chapitre contient également du matériel tiré des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER) :
“Caractéristiques fondamentales de l’eau“, une section de la chimie aquatique (par Chieh) des modules supplémentaires de Chemistry Libretextssur la chimie environnementale, utilisée sous licenceCC BY-NC-SA 3.0,
Paragraphes 17 et 18,
Section 7.6 “WritingChemical Equationsfor Reactionsin Solution- Molecular, Complete Ionic, and Net Ionic Equations“, une section de la carte de texte Chemistry Libretexts pour la chimie d’introduction (par Tro), utilisée sous une licence CC BY-NC-SA 3.0,
Paragraphes 24 à 29,
Exemples 1.4.14 et 1.4.16,
“Vérifiez votre apprentissage” 1.4.15 et 1.4.16, et
Section 16.11 “Molalité“, une section de la chimie d’introduction (CK-12), utilisée sous une licenceCC BY-NC 4.0,
Les paragraphes 14 à 16, et
Exemple 1.4.10.
Ce chapitre contient le contenu original du Dr. Brandi West, y compris la deuxième phrase du paragraphe 24.
Ce chapitre contient le contenu original de Jessica Thomas, y compris la phrase 2 du paragraphe 9, la phrase de l’exemple 1.4.6 au-dessus du sous-titre “Vérifiez votre apprentissage”, la question et la réponse pour (a) dans “Vérifiez votre apprentissage” 1.4.9, la dernière phrase du paragraphe 20, les 3 premiers points numérotés sous “Équations moléculaires, ioniques complètes et nettes” et les questions de fin de chapitre de 8 à 16.
Ce chapitre contient des réponses originales aux questions 7 à 18 créées par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient l’équation du point 1 sous “Équations moléculaires, ioniques complètes et nettes” (Réactions acide-base).
Ce chapitre contient l’équation du point 2 sous “Équations moléculaires, ioniques complètes et nettes” (5.6 – Réactions d’oxydation-réduction (Redox)).
Ce chapitre contient un exercice et sa réponse à la question 21 de la fin du chapitre, tirés de la section 4.2 “Classifier les réactions chimiques” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient des figures et du matériel provenant de (3.4 – Autres unités pour les concentrations de solutions), notamment les figures 1.4.3, 1.4.4, 1.4.5 et 1.4.7 et les paragraphes 6 et 8 à 13, la première phrase du paragraphe 1, les exemples 1.4.6, 1.4.7, 1.4.8 et 1.4.9, et “Vérifiez votre apprentissage” 1.4.6, 1.4.7, 1.4.8 et 1.4.9 (b).
Ce chapitre contient les figures 1.4.1, 1.4.2, et 1.4.6 tirées de “Molarité“.
Ce chapitre contient les figures 1.4.8 tirées de “Writing Chemical Equations for Reactions in Solution- Molecular, Complete Ionic, and Net Ionic Equations“.
1.5 – Réactions d’oxydoréduction
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 4.2 “Classification des réactions chimiques” et ses exercices,
Paragraphes 1-3 et 5-9,
Exemples 1.5.1 et 1.5.2,
“Vérifiez votre apprentissage” 1.5.1 et 1.5.2, et
Section 17.1 “Examen de la chimie redox” et ses exercices,
Exemples 1.5.3 et 1.5.5, et
“Vérifiez votre apprentissage” 1.5.4 et 1.5.5,
tous deux utilisés sous une licenceCC BY 4.0.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Phrases dans les étapes de l’exemple 1.5.5,
“Trucs et astuces – acronymes d’oxydation et de réduction”,
Au paragraphe 5 : le “(par exemple)” pour les points 1, 2 et 4, et la dernière partie de la phrase, sous le point 4, après la dernière virgule aux points 4.2 et 4.3,
Paragraphe 6,
Les phrases 1 et 5 du paragraphe 10, ainsi que les étapes 1 et 9, et
“Note” dans l’exemple 1.5.3 et la phrase dans l’exemple 1.5.3 concernant l’étape 7 de la solution de l’exemple.
Ce chapitre contient le contenu original de Geneviève O’Keefe, y compris la deuxième et dernière phrase du paragraphe 1.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient les réponses originales aux questions 1, 3, 5, 10 et 11 créées par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient la figure 1.5.2 tirée de “Classification des réactions chimiques“.
Chapitre 1 Termes clés
Les définitions des termes clés suivants ont été adaptées du chapitre 2 “Key Terms” du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Formule empirique
|
Isomères
|
Formule moléculaire
|
Formule structurelle
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 3 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Solution aqueuse
|
Dilution
|
Molarité (M)
|
Solute
|
|
Le numéro d’Avogadro (NA)
|
Masse de la formule empirique
|
Mole (n)
|
Solvant
|
|
Concentré
|
Pourcentage de masse (m/m %)
|
Parties par milliard (ppb)
|
Pourcentage en volume (v/v %)
|
|
Concentration (C)
|
Pourcentage masse-volume (m/v %)
|
Parties par million (ppm)
|
|
|
Diluer
|
Masse molaire (Mm)
|
Composition en pourcentage
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 4 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Rendement réel
|
Excès de réactif
|
Pourcentage de rendement
|
Spectateur
|
|
Équation équilibrée
|
Limiter le réactif
|
Réaction aux précipitations
|
Facteur stœchiométrique
|
|
Coefficient
|
Équation ionique nette
|
Produit
|
Stoechiométrie
|
|
Analyse de la combustion
|
Numéro d’oxydation
|
Réactif
|
Rendement théorique
|
|
Réaction de combustion
|
Réaction d’oxydation-réduction
|
Agent réducteur
|
|
|
Équation ionique complète
|
Agent oxydant
|
Réaction à un seul déplacement
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 11 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Amphipathique
|
Molalité (b)
|
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du glossaire du manuel ouvert Introductory Chemistry – 1st Canadian Edition (par Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0 :
|
Équation chimique
|
Diluer
|
Mole (n)
|
Réduction
|
|
Coefficient
|
Demi-réaction
|
Oxydation
|
Réaction à un seul déplacement
|
|
Réaction de combustion
|
Méthode de demi-réaction
|
Réaction d’oxydation-réduction
|
Solute
|
|
Concentration
|
Limiter le réactif
|
Pourcentage de rendement
|
Solvant
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées d’un autre manuel scolaire ouvert du projet LibreTexts de l’Open Education Resource (OER) :
Hydrophiles et hydrophobes – tiré de “Fundamental Characteristics of Water“, une section de Aquatic Chemistry (par Chieh) desmodules supplémentaires de Chemistry Libretextssur la chimie environnementale, utilisé sous licenceCC BY-NC-SA 3.0.
2 – Les gaz
2.1 – Forces intermoléculaires
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 10.1 “Forces intermoléculaires” et ses exercices,
Paragraphes 1 à 22,
Exemple 2.1.1,
“Vérifiez votre apprentissage” 2.1.1 – 2.13,
Figures 2.1.1 – 2.1.13, et
Questions et réponses 2, 5, 6 et 7.
Ce chapitre contient également du matériel tiré des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER) :
Section 3.7 “Forces intermoléculaires“, une section unitaire du cours CHEM1130 Principes de chimie I (du Northern Alberta Institute of Technology), utilisée sous licenceCC BY-NC-SA 3.0,
Paragraphe 23,
Questions 1, 3 et 4, et
Réponses 1, 2, 3, 4, 5, 6 et 7.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Phrase 3 du paragraphe 3,
La quatrième phrase du paragraphe 16, et
Réponse à la question “Vérifiez votre apprentissage” 2.1.3.
Ce chapitre contient du contenu original de Jessica Thomas, y compris :
Résumé à la fin du chapitre.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient les réponses originales aux questions 5 créées par Nathan Biniam et Leanne Trepanier.
2.2 – Les gaz et le tableau périodique
Ce chapitre contient des éléments tirés de la section 10.1 “Caractéristiques des gaz” de la carte de texte Libretextes sur la chimie : The Central Science (par Brown, LeMay, Busten, Murphy, et Woodward) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 4.0, y compris :
Paragraphes 1 à 3,
Exemples,
“Vérifiez votre apprentissage”, et
Figure 2.2.1.
Ce chapitre contient le contenu original de Jessica Thomas et du Dr. Kathy-Sarah Focsaneanu, y compris :
Questions et réponses 1 et 2.
2.3 – Mesure des variables des gaz
Ce chapitre contient du matériel et/ou des exemples tirés des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER) :
Section 2.2 “Expressing Units“, une section de Beginning Chemistry (by Ball), utilisée sous une licence CC BY-NC-SA 4.0,
Paragraphes 18-19,
Figure 2.3.5, et
Section 2.4 “Température“, une unité du cours CHM101 : Chemistry and Global Awareness (par Gordon de l’université Furman), utilisée sous licenceCC BY-NC-SA 4.0,
Paragraphes 20-26,
Exemple 2.3.6,
Figure 2.3.6, et
Section 5.2 “Gas Pressure and Its Measurement“, une section de la carte de texte de Chemistry Libretexts pour Chemistry – The Molecular Nature of Matter and Change (par Silberberg), utilisée sous une licence CC BY-NC-SA 3.0,
Paragraphes 8 à 16
Exemples 2.3.4, 2.3.5,
Figures 2.3.2, 2.3.3, 2.3.4,
“Vérifiez votre apprentissage” 2.3.3, 2.3.4, 2.3.5, et
Sections 9.3 “Pressure” et 9.4 “Measurementof Pressure”, sections de ChemPRIME (par Moore et al.), toutes deux utilisées sous une licenceCC BY-NC-SA 4.0,
Paragraphes 1 à 7,
Exemples 2.3.1, 2.3.3,
Figure 2.3.1,
“Vérifiez votre apprentissage” 2.3.2, et
Questions 8 et 9.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Les modifications apportées aux paragraphes 9, 12, 15, 18 et 23, et
Fin du chapitre questions 1, 2, 3, 5, 6 et 11.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris :
Réponses à la fin du chapitre.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Sous-rubrique de la figure 2.3.1.
Ce chapitre contient le contenu original de Leanne Trepanier et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
2.4 – Lois sur le gaz
Ce chapitre contient du matériel et des exercices tirés de la section 9.2 “RelatingPressure, Volume, Amount, and Temperature: The Ideal Gas Law” et ses exercices, respectivement, de la ressource de manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
Paragraphes 1 à 25,
Exemples 2.4.1 – 2.4.6,
Figures 2.4.1 – 2.4.10,
“Vérifiez votre apprentissage” 2.4.1 – 2.4.6,
Questions 1 à 14, et
Réponses 11 et 14.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Modifications des titres, des équations et des chiffres.
Ce chapitre contient un contenu original de Leanne Trepanier et Geneviève O’Keefe, y compris la numérotation des équations, des chiffres et les réponses 1, 3, 4, 5, 7, 9 et 10.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris les modifications apportées à la section Laboratoire CHEM1311.
Ce chapitre contient les réponses originales aux questions 2, 6, 8, 12 et 13 créées par Nathan Biniam.
2.5 – Mélanges de gaz et pressions partielles
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 9.3 “Stoechiométriedes substances, mélanges et réactions gazeuses” et ses exercices,
Paragraphes :
Sous l’équation 2.5.1 (paragraphe entier),
Paragraphe 4, deuxième phrase,
Paragraphe 5 (paragraphe entier),
Paragraphe 6 (paragraphe entier),
Paragraphe 7 (paragraphe entier),
Exemples,
Figure 2.5.1, et
“Vérifiez votre apprentissage”.
2.6 – Théorie cinétique moléculaire des gaz (comportements idéaux des gaz)
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 9.5 “La théorie cinétique et moléculaire” et ses exercices,
Paragraphes 1 à 12,
Exemple 2.6.1,
Figures 2.6.1, 2.6.2, 2.6.3, 2.6.4, 2.6.5, et
“Vérifiez votre apprentissage” 2.6.1.
Ce chapitre contient le contenu original de Leane Trapier et Derek Fraser-Halberg, y compris la numérotation des équations et les sous-titres des équations.
Ce chapitre contient du contenu original de Jessica Thomas, y compris :
Phrase 1 du paragraphe 9 (ajoutée “où M est la masse moyenne des particules”),
Paragraphe 10 (ajouté “(pour étendre votre apprentissage, consultez la dérivation pour le KEavgici) :”),
La première phrase de la réponse à “Vérifiez votre apprentissage” 2.6.1, et
Questions 1-8.
Ce chapitre contient du contenu original de Geneviève O’Keefe, notamment
Insérer “diminuer les deux” dans la loi de Charles – phrase 3, et
Réponses 2, 5 et 7.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Ajout de la “loi de Gay-Lussac” à “La théorie cinétique et moléculaire explique le comportement des gaz, partie I – Amontons”,
Ajout d’un paragraphe sous l’équation 2.6.2, et
Ajout de “exprimé en kilogrammes” au paragraphe 9.
Ce chapitre contient du contenu original de Nathan Biniam, notamment
Fin du chapitre réponses 1, 3, 4, 6 et 8.
2.7 – Diffusion et effusion
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 9.4 “Effusion et diffusion des gaz” et ses exercices,
Paragraphes 1 à 10,
Exemples 2.7.1, 2.7.2, 2.7.3,
Figures 2.7.1, 2.7.2, 2.7.3, et
“Vérifiez votre apprentissage” 2.7.1, 2.7.2, 2.73.
Ce chapitre contient du contenu original de Genevieve O’Keefe, notamment
Les deux premières phrases du paragraphe 1,
Paragraphe 6, première phrase,
Exemple 2.7.3,
Paragraphe 1, troisième phrase,
Solution – Phrase 4,
Un paragraphe a été ajouté au-dessus de “Vérifiez votre apprentissage” 2.7.3,
Il a écrit “Au cas où vous seriez intéressé… Utilisation de la diffusion pour les applications de l’énergie nucléaire : Section “Enrichissement de l’uranium”, et
Réponses aux questions 1, 3, 5, 7 et 9.
Ce chapitre contient du contenu original de Jessica Thomas, y compris :
Questions 1 à 9.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Ajouté à la phrase 1 de la section “La théorie cinétique et moléculaire explique le comportement des gaz, partie II,
Exemple 2.7.3 “Éléments et masses molaires”, et
Réponse à la question 6.
Ce chapitre contient du contenu original de Leanne Trepanier, notamment :
Réponses aux questions 2, 3, 4, 8, et
Passez aux questions 1 et 8.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Paragraphe 7,
Exemple 2.7.1,
Ajout de quelques mots au premier paragraphe, et
Solution complète (autre que l’équation).
Ce chapitre contient le contenu original de Leane Trapier et Derek Fraser-Halberg, y compris la numérotation des équations et les sous-titres des équations.
2.8 – Comportements réels ou non des gaz
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, notamment :
Section 9.6 “Comportement des gaz non toxiques” et ses exercices,
Paragraphes 1 à 9,
Exemple 2.8.1,
Tableau 2.8.1,
Figures 2.8.1, 2.8.2, 2.8.3, 2.8.4, et
“Vérifiez votre apprentissage” 2.8.1.
Ce chapitre contient du contenu original de Jessica Thomas, y compris :
Questions 1 à 7.
Ce chapitre contient du contenu original de Leanne Trepanier, notamment :
Réponse à la question 4, phrases 2 et 3.
Ce chapitre contient du contenu original de Geneviève O’Keefe, notamment
Réponse aux questions 1, 3, 5 et 7.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Phrase 3 du paragraphe sous la figure 2.8.3,
Figure 2.8.2 phrase 2, et
Petites modifications sur le paragraphe sous la figure 2.8.2.
Ce chapitre contient du contenu original de Nathan Biniam, notamment
Réponses aux questions 2, 4, 6
Ce chapitre contient le contenu original de Leane Trapier, Jessica Thomas et Derek Fraser-Halberg, y compris la numérotation des équations et les sous-titres des équations.
Chapitre 2 Termes clés
La définition du terme clé suivant a été adaptée du chapitre 1 “Key Terms” du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
Les définitions des termes clés suivants ont été adaptées du chapitre 9 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Zéro absolu
|
Diffusion
|
Manomètre
|
Atmosphère standard (atm)
|
|
La loi d’Avogadro
|
Effusion
|
Signifie chemin libre
|
Volume molaire standard
|
|
Bar (bar)
|
La loi Gay-Lussac
|
Fraction molaire (Χ)
|
Torr
|
|
Baromètre
|
La loi de l’effusion de Graham
|
Pression partielle
|
L’équation de Van der Waals
|
|
La loi de Boyle
|
Le gaz idéal
|
Pascal (Pa)
|
Pression de vapeur de l’eau
|
|
La loi de Charles
|
Constante de gaz idéale (R)
|
Pression (P)
|
|
|
Facteur de compressibilité (Z)
|
La loi idéale sur le gaz
|
Taux de diffusion
|
|
|
La loi des pressions partielles de Dalton
|
Théorie cinétique moléculaire
|
Vitesse quadratique moyenne (urms)
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 10 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Force de dispersion
|
Dipôle induit
|
Force intermoléculaire
|
La force de Van der Waals
|
|
Liaison hydrogène
|
Dipôle instantané
|
Polarisabilité
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du glossaire du manuel ouvert Introductory Chemistry – 1st Canadian Edition (par Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0 :
|
Zéro absolu
|
Force de dispersion
|
Torr
|
|
|
Facteur de compressibilité (Z)
|
Le gaz idéal
|
L’équation de Van der Waals
|
|
|
Attraction dipôle-dipôle
|
Théorie cinétique moléculaire
|
Vapeur
|
|
|
|
|
|
|
|
|
|
|
La définition du terme clé suivant a été adaptée d’un autre manuel scolaire ouvert du projet LibreTexts de l’Open Education Resource (OER) :
Pression atmosphérique – de ChemPRIME (par Moore et al.), utilisé sous une licenceCC BY-NC-SA 4.0.
3 – La thermochimie
3.1 – Introduction à la thermochimie
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 5.1 “Lesbases de l’énergie“,
Paragraphes 1 à 8,
Figure 3.1.1, et
Section 5.2 “La première loi de la thermodynamique“,
Les paragraphes 14-15, et
Figures 3.1.3.
Ce chapitre contient le contenu original de Leanne Trepanier, y compris les réponses aux questions 1, 2 et 4.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Figure 3.1.1 sous-titre,
A écrit des titres,
a rédigé la section “Mesurer les calories nutritionnelles”, et
Ajout de quelques mots à la dernière phrase du paragraphe 11.
Ce chapitre contient le contenu original du Dr. Brandi West, y compris :
Phrase 1 du paragraphe 1 de la section “L’énergie dans l’univers”,
Ajout de quelques mots aux questions 1 à 4, et
Rédaction d’une réponse aux questions 3 et 4.
Ce chapitre contient du contenu original de Geneviève O’Keefe, notamment
Ajout de quelques mots à la dernière phrase du paragraphe 11,
A écrit le titre et la première phrase de la section “L’énergie dans l’univers”,
“Mesurer les calories nutritionnelles” paragraphe 3 dernière phrase,
Les phrases 3 à 5 du paragraphe 7, et
Paragraphe 10.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris les questions 1 à 4.
3.2 – Types d’énergie
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Sections 5.1 “Notions debase sur l’énergie” et 5.3 “Enthalpie”.
Paragraphes 1, 2, 3 (dernière phrase), 4-7,
“Vérifiez votre apprentissage” 3.2.1,
Exemple 3.2.2,
Figures 3.2.1, 3.2.2, 3.2.3, 3.2.4,
“InternalEnergy“, une section de Thermodynamics (contribution d’Alborzfar) des modules complémentaires de Chemistry Libretextssur la chimie physique et théorique,
Paragraphes 3, phrases 1-2,
“Work and Heat“, une section du supplément de chimie générale (par Eames), utilisé sous une licenceCC BY 4.0,
Paragraphes 12, phrases 1 à 4 et phrase 13,
Section 1.2 “Lachaleur en tant que mécanisme de transfert d’énergie“, une unité du cours de Chimie générale 2B avec mention,
Paragraphes 8 à 10,
Section 5.4 “Enthalpie de réaction“, une section de la carte de texte des Libretextes de la chimie pour la chimie : La science centrale,
Paragraphes 12, phrases 5 à 8,
Section 7.2 “Travail et chaleur“, une section unitaire du cours hybride CHEM 051 – Principes fondamentaux de chimie I,
Le paragraphe 11, et
Exemple 3.2.1.
Ce chapitre contient le contenu original de Leanne Trepanier, y compris la numérotation des équations et la réponse à la question 5.
Ce chapitre contient le contenu original de Geneviève O’Keefe, y compris l’ajout de quelques mots aux questions 1 et 5.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris l’ajout de quelques mots aux questions 1.
Ce chapitre contient le contenu original de Nathan Biniam, y compris la numérotation des équations et les réponses aux questions 1 à 4.
3.3 – Première loi de la thermodynamique
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, notamment :
Section 5.3 “Enthalpie“,
Les paragraphes 1 (phrases 1-3), 3 (phrases 2-8), 4, 5, 7, et
Figures 3.3.1 et 3.3.3.
Ce chapitre contient du contenu original de Leanne Trepanier, notamment :
La numérotation des équations,
Questions 1, 2, 4 et 5, et
Réponses 1 à 4.
Ce chapitre contient le contenu original du Dr. Brandi West, y compris :
Paragraphe 1, dernière phrase,
Paragraphe 2,
Paragraphe 3, première phrase,
Changement de titres,
Paragraphe 6,
Options pour les questions 2 et 5,
Question 3 et 6, et
Réponses à la question 5.
3.4 – Enthalpie
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Sections 5.2 “Calorimétrie” et 5.3 “Enthalpie”, et ses exercices,
Paragraphes 1-6, 7 (de la section 5.3) 8-11,
Figure 3.4.1,
Exemples 3.4.1, 3.4.2,
“Vérifiez votre apprentissage” 3.4.1, 3.4.2,
Questions 1-8, et
Réponses 6 et 8.
Ce chapitre contient le contenu original de Leanne Trepanier, Mahdi Zeghal, Nathan Biniam et Derek Fraser-Halberg, y compris les équations de numérotation, les réponses et les sous-titres.
Ce chapitre contient le contenu original du Dr. Brandi West, y compris :
Question 3 et réponse.
3.5 – Calorimétrie
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Sections 5.1 “Lesbases de l’énergie“.
Paragraphes 3-7, 9 -11, 13-15,
Exemples 3.5.1, 3.5.2,
Figure 3.5.1,
“Vérifiez votre apprentissage” 3.5.2,
5.2 “Calorimétrie,”
Paragraphes 1 (phrases 1-4), 17-20, 21 (phrases 4, 5), 24-27, 29,
Exemples 3.5.2 – 3.5.7,
Figures 3.5.6, 3.5.8,
“Vérifiez votre apprentissage” 3.5.3 – 3.5.8,
Venkateswaran, R. Chimie générale,
Laboratoire CHM1311 | Expérience 2 : Enthalpie de diverses réactions, et
Exercices,
Questions 1 à 13.
Ce chapitre contient le contenu original de Leanne Trepanier et Nathan Biniam, y compris la numérotation des équations.
Ce chapitre contient du contenu original de Geneviève O’Keefe, notamment
Paragraphe 1, phrases 5 à 8,
Paragraphes 8 et 12,
Paragraphe 15, phrases 2 et 5,
Paragraphe 16, première phrase,
Sous-titres,
Paragraphe 20, phrases 1 à 3,
Paragraphe 21-22,
Ajout d’une phrase dans l’exemple de solution 3.5.4 et 3.5.5,
Ajout d’un paragraphe à la solution 3.5.6 “Vérifiez votre apprentissage”,
Ajout d’un paragraphe à la solution de l’exemple 3.5.6,
Est-ce que la section “Au cas où vous seriez intéressé… La thermochimie des chauffe-mains”
A fait la section “Calorimétrie des bombes – Vidéo”,
Exemple 3.5.7 Partie (b), et
Réponses aux questions 5, 7, 9 et 10.
Ce chapitre contient le contenu original du Dr. Brandi West, y compris :
Solution par exemple 3.5.7.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Tableau des chaleurs spécifiques des substances courantes à 25°C et 1 bar.
3.6 – Loi de Hess
Ce chapitre contient des éléments tirés des sections 5.1 “Energy Basics” et 5.3 “Enthalpy” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous une licenceCC BY 4.0, notamment :
Section 5.3 “Enthalpie” et ses exercices,
Paragraphes 2 à 15,
Exemples 3.6.1 – 3.6.7,
Figures 3.6.1 – 3.6.5,
“Vérifiez votre apprentissage” 3.6.1 – 3.6.7, et
Section 5.4 “Enthalpie de réaction” et “Changements d’enthalpie dans les réactions” de la carte de texte des Libretextes de chimie: The Central Science,
Paragraphe 1.
Ce chapitre contient le contenu original du Dr. Brandi West, y compris :
Ajout de la phrase 3 -6 au paragraphe 1,
Ajout de la phrase 3 sous la figure 3.6.1, et
Ajout de “Kilo – watts heures” à la question 20, partie (f).
Ce chapitre contient du contenu original de Leanne Trepanier, notamment :
Numérotation des équations.
Ce chapitre contient le contenu original de Leane Trapier, Nathan Biniam et le Dr. Kathy-Sarah Focsaneanu, y compris les questions de fin de chapitre.
Ce chapitre contient le contenu original de Nathan Biniam, y compris la fin de la réponse au chapitre (Geneviève O’Keefe a fait la réponse à la question 18) et le tableau 3.6.2.
Chapitre 3 Termes clés
Les définitions des termes clés suivants ont été adaptées du chapitre 5 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Calorimètre à bombe
|
Procédé exothermique
|
Joule (J)
|
Les alentours
|
|
Calorie (cal)
|
Travaux d’expansion
|
Énergie cinétique (Ek)
|
Système
|
|
Calorimètre
|
Première loi de la thermodynamique
|
Énergie potentielle (Epot)
|
Température (T)
|
|
Thermodynamique chimique
|
Chaleur (q)
|
Capacité thermique spécifique (c)
|
Énergie thermique
|
|
Processus endothermique
|
Capacité thermique (C)
|
Enthalpie standard de la combustion(ΔHc°)
|
Thermochimie
|
|
Énergie (E)
|
La loi de Hess
|
Enthalpie standard de formation(ΔHf°)
|
Travail (w)
|
|
Enthalpie (H)
|
Hydrocarbures
|
État standard
|
|
|
Changement d’enthalpie(ΔH)
|
Énergie interne (U)
|
Fonction de l’État
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du glossaire du manuel ouvert Introductory Chemistry – 1st Canadian Edition (par Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0 :
|
Calorimétrie
|
Température (T)
|
|
|
|
|
|
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées à partir d’autres ressources de manuels scolaires ouverts du projet LibreTexts de ressources éducatives ouvertes (REL) :
Système fermé, système isolé et système ouvert – de la section 5.2 “La première loi de la thermodynamique” de la carte de texte Libretextes pour la chimie : The Central Science (par Brown, LeMay, Busten, Murphy, et Woodward), utilisé sous une licenceCC BY-NC-SA 4.0, et
Path function – de “State vs. Path Functions“, une section de Fundamentals of Thermodynamics (contribué par Billings, Morris, Starr, et Oberoi) des modules supplémentaires de Chemistry Libretextssur la chimie physique et théorique, utilisés sous une licenceCC BY-NC-SA 3.0.
4 – Équilibre chimique
4.1 – Introduction à l’équilibre chimique
Ce chapitre contient du matériel et des exercices tirés de la section 13.1 “Équilibres chimiques” et de ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient également des informations tirées des manuels scolaires ouverts suivants :
Section 13.1 “Équilibres chimiques“, une section de chimie (par l’université du riz), utilisée sous licenceCC BY 4.0, et
Section 15.2 “L‘expression de la constante d’équilibre“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0, y compris :
Paragraphes 13 à 15, et la deuxième phrase du paragraphe 17,
Équations 4.1.1 et 4.1.2,
Exemple 4.1.1, et
“Vérifiez votre apprentissage” 4.1.1.
Ce chapitre comprend des éléments tirés de 13.1 – Équilibres chimiques, y compris les paragraphes 1-9 et 11, et l’encadré “Équilibre et boissons non alcoolisées”.
Ce chapitre comprend le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris la première phrase pour la réponse à la question 4 de la fin de la section 4.1 des questions.
Ce chapitre comprend des documents originaux de Mahdi Zeghal, notamment la phrase après le tiret du paragraphe 1, les paragraphes 10 et 16, la première et dernière phrase du paragraphe 11, la “note” sous l’équation 4.1.2, le tableau 4.1.1, les deux dernières phrases du paragraphe 17 et la case “quand dois-je utiliser une flèche à un côté ?
Ce chapitre comprend des documents originaux de Leanne Trepanier et Nathan Biniam, y compris la réponse à la fin de la section 4.1, questions 2 et 4.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des figures, des tableaux et des équations.
Ce chapitre comprend les figures 4.1.1, 4.1.2, 4.1.3 et 4.1.4 tirées de la section 13.1 – Équilibres chimiques.
Ce chapitre comprend la figure 4.1.5 tirée de “The Equilibrium Constant Expression“.
4.2 – La constante d’équilibre et le quotient de réaction
Ce chapitre contient du matériel et des exercices tirés de la section 13.2 “EquilibriumConstants” et ses exercices, respectivement, de la ressource de manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
Paragraphe 3.
Ce chapitre contient également des informations tirées des sections 15.2 “L‘expression de la constante d’équilibre“, 15.3 “Relations impliquant les constantes d’équilibre” et 15.5 “Le quotient de réaction, Q – Prédire la direction du changement net” de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0, y compris :
Section 15.2 :
Paragraphes 2 et 5 à 9,
Équations 4.2.1, 4.2.2 et 4.2.3,
Exemple 4.2.1,
“Vérifiez votre apprentissage” 4.2.1 et 4.2.2,
Section 15.3 :
Paragraphes 10 à 18,
“Résumé” au-dessus de l’exemple 4.2.2,
Équations 4.2.4 et 4.2.5,
Exemples 4.2.2, 4.2.3 et 4.2.5,
“Vérifiez votre apprentissage” 4.2.3 et 4.2.5, et
Section 15.5 :
Les paragraphes 19-20 et 22-23, et
Équation 4.2.8.
Ce chapitre contient des éléments tirés de la section 13.2 – Constantes d’équilibre, y compris :
Les puces du paragraphe 3 et la section de l’équation 4.2.6 écrite ci-dessous,
Équations 4.2.6 et 4.2.7,
Exemples 4.2.4, 4.2.6, 4.2.7 et 4.2.8, et
“Vérifiez votre apprentissage” 4.2.4, 4.2.6, 4.2.7 et 4.2.8.
Ce chapitre comprend les questions de la fin de la section 4.2 et ses réponses tirées de la section 13.2 “Equilibrium Constants” du manuel ouvertChemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre comprend des documents originaux du Dr. Kathy-Sarah Focsaneanu, dont
La partie de l’écriture après le “et” dans la phrase 3 du paragraphe 2,
La deuxième phrase du troisième point du paragraphe 3,
“Pour plus d’informations sur les activités, cliquez ici“,
La deuxième phrase sous la solution pour (a) et (d) de l’exemple 4.2.1,
La dernière phrase du paragraphe 15,
La deuxième phrase du paragraphe 16,
La description du PK et du R selon l’équation 4.2.6,
La deuxième phrase dans la case marquée sous l’équation 4.2.7, et
La dernière phrase du paragraphe 21.
Ce chapitre comprend des documents originaux de Mahdi Zeghal, dont
Les paragraphes 1, 4, 21 ainsi que les puces,
Le troisième point et la dernière partie de la phrase après la virgule du point 4 du paragraphe 3,
“Note” sous le paragraphe 4,
La dernière phrase du paragraphe 7,
Les parenthèses du paragraphe 13,
Sous “résumé”, les parenthèses des points 1 et 2, ainsi que la totalité du troisième point,
La première phrase de la case marquée sous l’équation 4.2.7,
“Note” sous la case marquée, et
La boîte “CHM 1311 Pointers”.
Ce chapitre comprend des documents originaux de Leanne Trepanier et Nathan Biniam, y compris les réponses aux questions 2, 3, 5, 10, 11, 12, 15 et 17 de la fin de la section 4.2.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient la figure 4.2.1 tirée de “Le quotient de réaction, Q – Prévoir la direction du changement net“.
4.3 – Résoudre les problèmes d’équilibre
Ce chapitre contient des éléments tirés de la section 13.4 “Equilibrium Calculations” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient également du matériel et des exercices tirés de la section 15.7 “Equilibrium Calculations– SomeIllustrative Examples” de la carte de texte Libretexts pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0.
Exemples 4.3.3 et 4.3.6, et
“Vérifiez votre apprentissage” 4.3.3 et 4.3.7.
Ce chapitre contient des éléments tirés de la section 13.4 – Calculs à l’équilibre, y compris :
Les paragraphes 1 à 9, 10 et les points en dessous, 11 à 12, 15 à 16 et 18 à 23,
Exemples 4.3.1, 4.3.2, 4.3.4, 4.3.5, 4.3.7 et 4.3.8, et
“Vérifiez votre apprentissage” 4.3.1, 4.3.2, 4.3.4, 4.3.5, 4.3.6, 4.3.8 et 4.3.9.
Ce chapitre contient le paragraphe 14 tiré de La constante d’équilibre.
Ce chapitre contient la fin de la section 4.3 questions 1-17 tirées de 15.3 – Résolution des problèmes d’équilibre.
Ce chapitre contient des documents originaux du Dr. Kathy-Sarah Focsaneanu, y compris le texte ci-dessus sur le “calcul d’une constante d’équilibre”.
Ce chapitre contient des documents originaux de Mahdi Zeghal, dont
Paragraphes 13 et 17,
“Notez” ci-dessous la solution de l’exemple 4.3.5,
Le début de la troisième phrase, avant les points-virgules, du paragraphe 22,
“Note” en dessous du paragraphe 23,
Cases marquées sous la rubrique “Vérifiez votre apprentissage” 4.3.8, et
La deuxième phrase, la partie après le tiret dans la cinquième phrase, la septième à la neuvième phrase et la partie après le tiret dans la dixième phrase dans la solution par exemple 4.3.8.
Ce chapitre comprend des documents originaux de Leanne Trepanier et Nathan Biniam, y compris les réponses à la fin de la section 4.3, questions 1 à 17.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres.
Ce chapitre contient la figure 4.3.2 tirée de “Equilibrium Calculations– SomeIllustrative Examples“.
4.4 – Le principe du Châtelier
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, notamment :
Section 12.7 “Catalyse“.
Le paragraphe 24, et
Section 13.3 “Changement des équilibres” : Le principe du Châtelier” et ses exercices,
tous utilisés sous une licenceCC BY 4.0.
Ce chapitre contient également du matériel et/ou des exemples tirés des manuels scolaires ouverts suivants :
Section 13.3 “Changement des équilibres” : Le principe du Châtelier“, une section de chimie (par l’Université du riz), utilisée sous une licenceCC BY 4.0,
Paragraphes 1-16 et 25-32,
“Effet du changement de pression sur l’équilibre – Démonstration vidéo”,
Tableau 4.4.1,
Section 14.E “Principles of Chemical Equilibria (Exercices)“, exercices de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0,
Section 19.2 “Le concept d’entropie“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0,
Section 19.7“ΔG° and K as Functions of Temperature“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0,
Paragraphes 20 à 23,
Équation 4.4.1,
Exemple 4.4.1,
“Vérifiez votre apprentissage” 4.4.1, et
Section 95 “Changement des équilibres” : Le Principe du Châtelier“, une section de l’introduction à la chimie – 1ère édition canadienne (par Key and Ball), utilisée sous licenceCC BY-NC-SA 4.0,
La boîte “Au cas où vous seriez intéressé… les équilibres dans le jardin”.
Ce chapitre contient la fin de la section 4.4 questions 1-16 et ses réponses tirées de la section 13.3 “Changement des équilibres” : Le Châtelier’s Principle” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient des documents originaux écrits par le Dr. Kathy-Sarah Focsaneanu, y compris les crochets de la deuxième phrase du paragraphe 18.
Ce chapitre comprend des documents originaux écrits par Mahdi Zeghal, dont
Paragraphes 17-19 et 33,
Les points de l’équation 4.4.1,
Première et dernière phrase du paragraphe 23,
“Note” en dessous du paragraphe 23,
“Note” au paragraphe 24,
Les deux premières phrases du paragraphe 25,
Boîte signalée sous le paragraphe 25, et
Dernière phrase de la case “Au cas où vous seriez intéressé… les équilibres dans le jardin”.
Ce chapitre contient des documents originaux de Leanne Trepanier et Nathan Biniam, y compris les réponses aux questions 4, 8, 9, 11, 12, 14, 15 et 16 de la fin de la section 4.4.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des figures, des tableaux, des équations et des exemples.
Ce chapitre contient les figures 4.4.1 et 4.4.3 tirées de “Shifting Equilibria” : Le principe du Châtelier,”.
Ce chapitre contient la figure 4.4.2 tirée de“ΔG° et K comme fonctions de la température,”.
Ce chapitre contient la figure 4.4.4 tirée de “Shifting Equilibria” : Le principe du Châtelier,”.
Chapitre 4 Termes clés
Les définitions des termes clés suivants ont été adaptées du chapitre 13 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Equilibre
|
Équilibre hétérogène
|
Le principe du Châtelier
|
Réaction réversible
|
|
Constante d’équilibre (K)
|
Équilibre homogène
|
Quotient de réaction (Q)
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du glossaire du manuel ouvert Introductory Chemistry – 1st Canadian Edition (par Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0 :
|
Entropie (S)
|
Constante d’équilibre (K)
|
|
|
|
|
|
|
|
|
|
|
|
La définition du terme clé suivant a été adaptée d’un autre manuel scolaire ouvert du projet LibreTexts de l’Open Education Resource (OER) :
Équation de van’t Hoff – de la section 19.7“ΔG° et K comme fonctions de la température“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.), utilisé sous une licenceCC BY-NC-SA 3.0.
5 – Équilibres acide/base
5.1 – Définitions des acides de base et paires d’acides de base conjugués
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 14.1 “Brønsted-Lowry Acids and Bases” et ses exercices
Paragraphes 12, 16-18,
Questions 1 à 7, et
Section 16.1 “Théorie d’Arrhenius : Un bref aperçu“.
Les paragraphes 1 à 11, et
Figure 5.1.1.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Ajout de la phrase 3 au paragraphe 18, et
Fin du chapitre réponses 2, 3, 4 et 7.
Ce chapitre contient du contenu original de Leanne Trepanier, notamment :
Réponses aux questions 1 et 6.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris l’étiquetage des chiffres et des équations, la rédaction de la réponse à la question 5 et l’aide à la rédaction de la question 7.
Ce chapitre contient du contenu par le Dr. Kathy-Sarah Focsaneanu, y compris :
Paragraphe 6, phrases 1 à 3,
Figure 5.1.1 sous-titre,
Paragraphe 4, phrases 1 à 3,
Ajout de “dissocie (rupture)” au paragraphe 7, première phrase,
Ajout d’une équation au paragraphe 8,
Ajout de “lorsqu’ils sont dissous dans l’eau” à la fin du paragraphe 9,
Ajout de la phrase 1 au paragraphe 10,
Ajout de quelques mots aux phrases 2 et 4 du paragraphe 10,
Rédaction du paragraphe 11,
Beaucoup de choses ont été ajoutées au paragraphe 12,
Ajouté à la première et dernière phrase du paragraphe 13,
A écrit le paragraphe 14 avec Brandi,
A écrit les phrases 1, 2, 3, 5 au paragraphe 15,
Ajout de quelques mots aux phrases 1 et 4 du paragraphe 18,
Ajouté à la première phrase du paragraphe 19, et
Il a rédigé les paragraphes 22 et 23.
Ce chapitre contient du contenu original de Geneviève O’Keefe, notamment
Ajout de quelques mots à la phrase 4 du paragraphe 15.
Ce chapitre contient le contenu original du Dr. Brandi West, y compris :
Rédaction du paragraphe 9,
Ajouté à la première et dernière phrase du paragraphe 13,
A écrit la dernière phrase du paragraphe 14,
Ajout de quelques phrases (1, 2, 3, 5) au paragraphe 15,
Il a rédigé les paragraphes 16 et 17,
A écrit la phrase 1-4 au paragraphe 18, et
Rédaction du paragraphe 21.
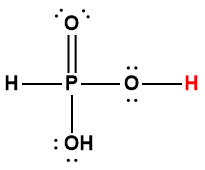
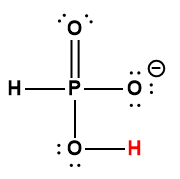
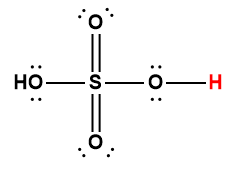
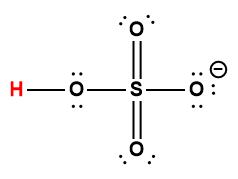
5.2 – Autoionisation de l’eau et pH/pOH
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Sections 14.1 “Brønsted-Lowry Acidsand Bases” et 14.2 “pH et pOH“, et ses exercices,
Paragraphes 1-21,
Figures 5.2.1 – 5.2.5,
Exemples 5.2.1 – 5.2.5,
“Vérifiez votre apprentissage” 5.2.1 – 5.2.5,
Questions 4, 6, 7, 8, 9, 10, 11, 12, 13, 14, et
Réponses 4, 6, 8, 10, 12, 14.
Ce chapitre comprend des documents originaux de Leanne Trepanier, Mahdi Zeghal et Derek Fraser-Halberg, notamment :
La numérotation des équations,
Sous-rubriques, et
Leanne Trepanier et Derek Fraser-Halberg ont rédigé les questions 1, 2, 3 et 5 de la fin du chapitre, ainsi que les réponses aux questions 1 à 3.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Rédaction du paragraphe 8,
Ajout de quelques mots à la phrase 1 du paragraphe 10,
A écrit le paragraphe au-dessus de la figure 5.2.3, et
A écrit la deuxième phrase du paragraphe 20.
Ce chapitre contient du contenu original de Geneviève O’Keefe, notamment
Ajout de quelques mots à la solution, par exemple 5.2.2,
Il a rédigé les équations entre les paragraphes 2 et 3, et
Ajout de quelques mots au paragraphe 21.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Ajout de quelques mots à la première et à la dernière phrase du paragraphe 1,
Les options ont été ajoutées à l’exemple 5.2.3,
Ajout de “tels que ceux figurant dans le tableau 5.2.1 ci-dessous” au paragraphe 14, et
Ajout de “solutions (pH > 7)” à la dernière phrase du paragraphe 15.
Ce chapitre contient le contenu original du Dr. Brandi West, y compris :
Rédaction du paragraphe 1,
A écrit la réponse à la question 5.2.1 “Vérifiez votre apprentissage”, et
A écrit le paragraphe 15 avec le Dr. Kathy-Sarah Focsaneanu.
5.3 – Force acide/base
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 14.3 “Forces relatives des acides et des bases” et ses exercices,
Paragraphes 1-50,
Figures 5.3.1 – 5.3.9,
“Vérifiez votre apprentissage” 5.3.1 – 5.3.9,
Exemples 5.3.1 – 5.3.9,
Questions 1 à 24, et
Réponses 1, 2, 6, 9, 11, 12, 14, 15, 17, 19, 21, 23, 24, 25, 27, 28, 30, 31, 32 et 34.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris la numérotation des équations et l’étiquetage des chiffres.
Ce chapitre contient du contenu original de Geneviève O’Keefe, notamment
Le paragraphe 5, et
Réponse à la question 16.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Paragraphe 2, phrases 3 et 4,
Paragraphe 3 phrase 2,
Sous-rubrique de la figure 5.3.2,
Paragraphe 9,
La majeure partie du paragraphe 13, y compris le titre “Essayez-le vous-même – Pourcentage d’ionisation et [H3O+]eq par rapport à la concentration initiale d’acide”,
Exemple 5.3.6 solution phrase 3,
Tableau 5.3.1, et
Ajout de quelques mots à la phrase 2 pour la solution “Vérifiez votre apprentissage” 5.3.2.
5.4 – Acides polyprotiques
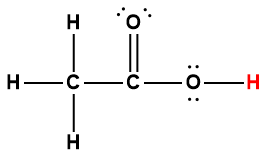
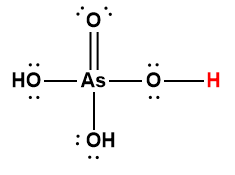
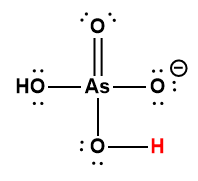
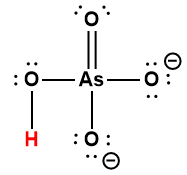
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 14.5 “Acides polyprotiques” et ses exercices,
Paragraphes 1-21,
Exemple 5.4.1,
Questions 1 à 5,
“Vérifiez votre apprentissage” 5.4.1 – 5.4.2, et
Un exemple/exercice tiré de “Solutions des systèmes acides/bases polyprotiques, problème D” du thème Chimie acide-base,
Exemple 5.4.2.
Ce chapitre contient du contenu original de Leanne Trepanier, notamment :
La numérotation des équations, et
Réponses aux questions 3 et 5.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris :
Il a rédigé les paragraphes 4 et 10, et
Création d’un tableau au-dessus de l’exemple 5.4.1.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Ajout de quelques mots au paragraphe 2,
Ajout de deux phrases au paragraphe 5,
A apporté des modifications à la première et à la dernière phrase du paragraphe 10,
A apporté de légères modifications aux noms des tables,
Ajout de quelques mots à la deuxième phrase de la réponse à la question 5.4.1 “Vérifiez votre apprentissage”, et
Ajout d’une équation à la troisième ionisation.
Ce chapitre contient le contenu original de Geneviève O’Keefe, y compris les réponses à la première question.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris la numérotation des équations et le sous-titre.
Ce chapitre contient un contenu original de Nathan Biniam, y compris la réponse à la question 5.4.2 “Vérifiez votre apprentissage” et les réponses aux questions 2 et 4.
5.5 – Hydrolyse des solutions salines
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 14.4 “Hydrolyse des sels” et ses exercices,
Exemples 5.5.1 – 5.5.3,
“Vérifiez votre apprentissage” 5.5.1 – 5.5.2,
Figure 5.5.2,
Section 12.5 “Acides et bases forts et faibles et leurs sels“, une section de la chimie debase,
Le paragraphe 2, et
Section 14.4 “Hydrolyse des solutions salines“.
Paragraphes
1, 3, 4, 5, 6, 7, et
les deux premiers paragraphes sous le titre “L’ionisation des ions métalliques hydratés”,
Figure 5.5.3, 5.5.1,
Exemple 5.5.5, et
“Vérifiez votre apprentissage” 5.5.5.
Ce chapitre contient le contenu original de Leanne Trepanier et Derek Fraser-Halberg, y compris la numérotation des équations.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
A écrit la phrase 5 au paragraphe 3,
A écrit la dernière phrase du paragraphe 15,
Exemple 5.5.3 phrases 7 et 8,
“Vérifiez votre apprentissage” 5.5.3,
A écrit la première phrase de la section “L’ionisation des ions hydratés”, et
Ajout de quelques mots à la première phrase du deuxième paragraphe dans la section “L’ionisation des ions hydratés”.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Rédaction des phrases 6 et 7 du paragraphe 3.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Nathan Biniam, y compris les réponses aux questions.
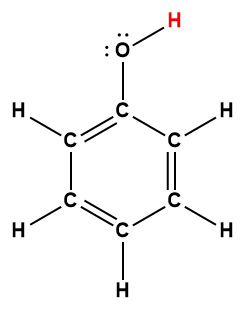
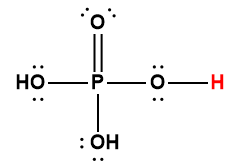
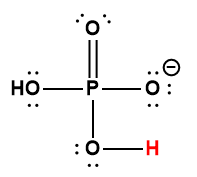
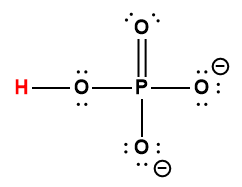
Ce chapitre contient du matériel et des exercices tirés des sections suivantes du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD :
Section 15.2 “Acides et bases de Lewis” et ses exercices,
Paragraphes 1-21,
Questions 1 à 16,
Section 15.2 “Acides et bases de Lewis,”
“Vérifiez votre apprentissage” 5.6.2,
Exemple 5.6.2, et
Section 16.9 “Acides et bases de Lewis“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principes et applications modernes,
Exemple 5.6.1, et
“Vérifiez votre apprentissage” 5.6.1.
Ce chapitre contient du contenu original de Leanne Trepanier, notamment :
La numérotation des équations,
Options pour la question 11, et
Réponses aux questions 2, 4 et 15 partie B.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris :
La numérotation des équations,
Options pour les questions 12 et 13,
Ajout de quelques mots pour la réponse 9, et
A écrit la réponse aux questions 14 et un peu de 15.
Ce chapitre contient le contenu original du Dr. Kathy-Sarah Focsaneanu, y compris :
Premier paragraphe,
Paragraphe 6, première phrase,
Réponse à “Vérifiez votre apprentissage” 5.6.1, paragraphe 1 phrase 1,
Paragraphe 6, deuxième phrase, et
Exemple 5.6.2 solution phrase 4, 7, 8 et 9.
Ce chapitre contient du contenu original de Geneviève O’Keefe, notamment
Rédaction de la solution 5.6.1 “Vérifiez votre apprentissage”,
Ajout de quelques mots à la dernière phrase du paragraphe 3, et
Réponses aux questions 1, 3, 5, 6, 7, 8, 9 et 13.
Chapitre 5 Termes clés
Les définitions des termes clés suivants ont été adaptées du chapitre 4 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Acide fort
|
Une base solide
|
Acide faible
|
Une base faible
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 14 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Ionisation de l’acide
|
Ionisation des bases
|
Base diprotique
|
Acide triprotique
|
|
Constante d’ionisation de l’acide (Ka)
|
Constante d’ionisation de la base (Kb)
|
Constante de produit ionique pour l’eau (KW)
|
pH
|
|
Acidique
|
De base
|
Effet de nivellement de l’eau
|
pOH
|
|
Amphiprotique
|
Acide conjugué
|
Acide monoprotique
|
|
|
Amphotère
|
Base conjuguée
|
Pourcentage d’ionisation
|
|
|
Autoionisation
|
Acide diprotique
|
Ionisation progressive
|
|
|
|
|
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 15 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Coordonner le lien covalent
|
Constante de formation (Kf)
|
Adduit de Lewis à base d’acide
|
La base de Lewis
|
|
Constante de dissociation (Kd)
|
Acide de Lewis
|
La chimie acido-basique de Lewis
|
Ligand
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du glossaire du manuel ouvert Introductory Chemistry – 1st Canadian Edition (par Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0 :
|
Amphiprotique
|
Autoionisation
|
Ion d’hydronium (H3O+)
|
pH
|
|
Acide d’Arrhenius
|
Acide de Brønsted-Lowry
|
Constante de produit ionique pour l’eau (KW)
|
pOH
|
|
Base d’Arrhenius
|
Base de Brønsted-Lowry
|
Réaction de neutralisation
|
|
|
|
|
|
6 – Tampons et titrages (équilibres ioniques dans les systèmes aqueux)
6.1 – Effet d’ions communs
Ce chapitre contient du matériel et des exercices tirés des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER), notamment
Section 17.1 “Common-Ion Effect in Acid-Base Equilibria“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.), utilisé sous une licenceCC BY-NC-SA 3.0,
Paragraphes 1-2 et 7-9,
Exemples 6.1.1 et 6.1.2, et
Section 17.E “Exercices“, exercices de la carte de texte des Libretextes de Chimie pour la Chimie Générale : Principles and Modern Applications (par Petrucci et al.), utilisé sous une licenceCC BY-NC-SA 3.0, comprenant
fin de la section questions 1 – 6 et ses réponses.
Ce chapitre contient du matériel tiré du Dr. Kathy Sarah-Focsaneanu, notamment
La troisième et la quatrième phrase du paragraphe 2,
Les phrases descriptives dans la solution de l’exemple 6.1.1,
Paragraphes 3 à 6,
La dernière phrase du paragraphe 7,
Exemples 6.1.3 et 6.1.4,
Les légendes des figures 6.1.1 et 6.1.2,
Le texte descriptif ci-dessus figure 6.1.1,
La première phrase du paragraphe 9, et
“Vérifiez votre apprentissage” 6.1.4.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des exemples.
Ce chapitre contient les figures 6.1.1 et 6.1.2 tirées de “Common-Ion Effect in Acid-Base Equilibria“.
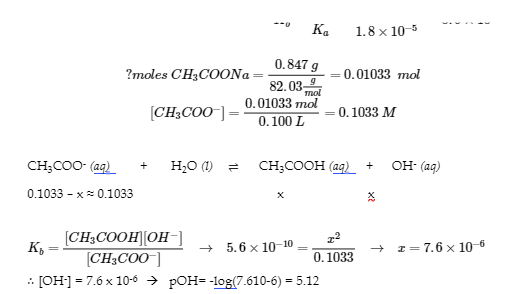
6.2 – Solutions tampons
Ce chapitre contient des éléments tirés de la section 14.6 “Buffers” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous une licenceCC BY 4.0, notamment :
Points 1 et 2 sous “Sélection de mélanges tampons appropriés”.
Paragraphes 6, 11 et 12,
L’équation Henderson-Hasselbalch” et la “médecine” : Système tampon dans le sang”, et
Boîte “Lawrence Joseph Henderson et Karl Albert Hasselbalch”.
Ce chapitre contient également du matériel tiré des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER) :
Section 17.2 “BufferedSolutions“, une section de la carte de texte des Libretextes de lachimie : The Central Science (par Brown, LeMay, Busten, Murphy et Woodward), utilisé sous une licence CC BY-NC-SA 4.0,
Les paragraphes 13, et
“Introduction to Buffers“, une section de Buffers (contribué par Pietri et Land) des modules supplémentaires de Chemistry Libretextssur la chimie physique et théorique, utilisé sous une licenceCC BY-NC-SA 3.0.
Ce chapitre contient du matériel tiré de Buffers, notamment :
Paragraphes 1 et 7 à 9,
Exemple 6.2.1, et
“Vérifiez votre apprentissage” 6.2.1.
Ce chapitre contient des documents originaux du Dr. Kathy-Sarah Focsaneanu, dont
Les flous sous “Comment fonctionnent les tampons” et au-dessus “Tampons acides : mélanges aqueux de HA + A-“,
Paragraphes 2 et 10,
La dernière phrase du paragraphe 5,
c) dans l’exemple 6.2.1,
La première phrase de la solution (b) dans l’exemple 6.2.1, et
Les 2 premières bavures sous “Médecine : le système tampon dans le sang”.
Ce chapitre contient des documents originaux de Jessica Thomas, notamment les paragraphes 3 à 5 et les “Courbes d’action tampon”.
Ce chapitre contient des documents originaux de Geneviève O’Keefe, y compris la “note” de l’exemple 6.2.1 pour la solution (a) du point 4.
Ce chapitre contient les figures 6.2.1, 6.2.2 et 6.2.3 tirées de “Buffers“.
Ce chapitre contient la figure 6.2.4 tirée de “BufferedSolutions“.
6.3 – Réactions acide-base et titrages
Ce chapitre contient du matériel tiré des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER) :
Section 14.10 “Titration Curves“, une section de ChemPRIME (par Moore et al.), utilisée sous une licenceCC BY-NC-SA 4.0,
La case “Quand une titration est-elle terminée”,
Section 17.3 “Titrages acide-base“, une section de la carte de textes Libretexts pour la chimie : The Central Science (par Brown, LeMay, Busten, Murphy et Woodward), utilisé sous une licenceCC BY-NC-SA 4.0,
Paragraphes 1, 18-20, 22-31 et 33-46,
Exemples 6.3.1 et 6.3.2,
“Vérifiez votre apprentissage” 6.3.1, 6.3.2 et 6.3.3,
Sections 17.3 “Indicateurs acide-base” et 17.4 “Réactions de neutralisation et courbes de titrage“, sections de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.), toutes deux utilisées sous licence CC BY-NC-SA 3.0,
Les paragraphes 2, 4 à 17, et
“pH and Food Color“, une section de “Foods” (contribution de Vitz et al.) des matériaux auxiliaires de Chemistry Libretexts, utilisés sous une licenceCC BY-NC-SA 3.0,
Tableau 6.3.1.
Ce chapitre contient du matériel original écrit par le Dr. Kathy-Sarah Focsaneanu, notamment
Paragraphes 3 et 21,
Phrases 3 à 6 du paragraphe 12,
La deuxième et la troisième phrase du paragraphe 14,
La deuxième phrase du paragraphe 18,
La deuxième et la troisième phrase du paragraphe 19,
Les phrases commençant après la cendre dans les phrases 2 à 3 du paragraphe 23,
Première et dernière phrase de 24,
La phrase ci-dessus “Calcul du pH pendant le titrage”,
La case “flèches d’équilibre”,
Toutes les boîtes BAMA,
La deuxième phrase du paragraphe 32,
Le paragraphe 2 de la solution par exemple 6.3.2,
“Vérifiez votre apprentissage” (a) 6.3.2,
La première phrase du paragraphe 37, et
Les parenthèses de la première phrase du paragraphe 38.
Il contient des documents originaux écrits par Jessica Thomas, notamment la première phrase de la case “flèches d’équilibre” et la première phrase du paragraphe 32.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des exemples, des figures et des tableaux.
Ce chapitre contient la figure 6.3.1 tirée de “pH et couleur des aliments“.
Ce chapitre contient les figures 6.3.2, 6.3.3 et 6.3.4 tirées de “Acid-Base Indicators“.
Ce chapitre contient les figures 6.3.5, 6.3.6, 6.3.7 et 6.3.8 tirées de “Titrages acide-base“.
6.4 – Équilibres des composés ioniques légèrement solubles
Ce chapitre contient du matériel et des exercices tirés de la section 15.1 “Précipitation et dissolution” et de ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0, y compris :
Fin du chapitre 6.4 questions 1-11 et ses réponses.
Ce chapitre contient également des informations tirées de la section 18.1 “SolubilityProduct Constant, Ksp” de la carte de texte Libretexts de chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0, y compris :
Paragraphes 10 à 12.
“SolubilityRules“, une section de Solubility (contribué par Mursa et Busch) des modules supplémentaires de Chemistry Libretextssur la chimie physique et théorique, utilisé sous une licenceCC BY-NC-SA 3.0, incluant :
Les paragraphes 1 à 3, et
Points soulevés dans la rubrique “Règles de solubilité”.
Ce chapitre contient des informations tirées du chapitre 15.1 Précipitation et dissolution – Chimie, y compris :
Paragraphes 4-9, 13-16, et 19-26,
Exemples 6.4.1, 6.4.2, 6.4.3, 6.4.4, 6.4.5, 6.4.6, 6.4.8, 6.4.9, 6.4.10 et 6.4.11,
“Vérifiez votre apprentissage” 6.4.1, 6.4.2, 6.4.3, 6.4.4, 6.4.5, 6.4.6, 6.4.10, 6.4.11, 6.4.12 et 6.4.13,
Tableau 6.4.1,
L’équation 6.4.1, et
Les encadrés “Utilisation du sulfate de baryum pour l’imagerie médicale” et “Le rôle des précipitations dans le traitement des eaux usées”.
Ce chapitre contient l’exemple 6.4.7 tiré de 18.3 : Effet des ions communs dans les équilibres de solubilité.
Ce chapitre contient “Check your learning” 6.7.8 tiré de 17.4 : Solubility Equilibria.
Ce chapitre contient des documents originaux du Dr. Kathy-Sarah Focsaneanu, dont
La première phrase du paragraphe 4,
La partie de la phrase qui suit les mots en italique au paragraphe 5,
Paragraphe 17,
Tout ce qui se trouve sous le sous-titre “Effet du pH sur la solubilité”, y compris “Vérifiez votre apprentissage” 6.4.9, et
Le premier paragraphe sous la solution de l’exemple 6.4.11.
Ce chapitre contient des réponses originales pour la fin de la section 6.4 questions 2, 3 et 6 créées par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient des réponses originales pour la fin de la section 6.4 question 10 créée par Geneviève O’Keefe.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des exemples, des figures, des tableaux et des équations.
Ce chapitre contient les figures 6.4.1, 6.4.3, 6.4.4 et 6.4.5 tirées de la section 15.1 Précipitation et dissolution – Chimie.
Ce chapitre contient la figure 6.4.2 tirée de “SolubilityProduct Constant, Ksp.
Chapitre 6 Termes clés
Les définitions des termes clés suivants ont été adaptées du chapitre 4 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Analyte
|
Point de fin (Endpoint)
|
Précipiter
|
|
|
Buret
|
Point d’équivalence
|
Titrant
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 11 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Saturée
|
Solubilité
|
Sursaturé
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 14 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Indicateur acide-base
|
Capacité tampon
|
Équation de Henderson-Hasselbalch
|
|
|
Tampon
|
Intervalle de changement de couleur
|
Courbe de titrage
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 15 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Effet d’ion commun
|
Solubilité molaire
|
Précipitations sélectives
|
Constante de produit de solubilité (Ksp)
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du glossaire du manuel ouvert Introductory Chemistry – 1st Canadian Edition (par Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0 :
|
Indicateur acide-base
|
Tampon
|
Buret
|
|
|
Analyte
|
Capacité tampon
|
Sursaturé
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées à partir d’autres ressources de manuels scolaires ouverts du projet LibreTexts de ressources éducatives ouvertes (REL) :
Produit ionique (Q) – de la section 18.1 “SolubilityProduct Constant, Ksp” de la carte de texte Libretexts de chimie générale : Principles and Modern Applications (par Petrucci et al.), utilisé sous une licenceCC BY-NC-SA 3.0, et
Point médian – de la section 17.3 “Titrages acide-base“, une section de la carte de texte Libretextes de la chimie pour la chimie : The Central Science (par Brown, LeMay, Busten, Murphy et Woodward), utilisé sous une licenceCC BY-NC-SA 4.0.
Fin du chapitre 6 Questions
Ce chapitre contient les questions 1 à 4 traduites et les solutions des examens passés du Dr. Alain St-Amant, dont la permission a été accordée.
7 – Cinétique chimique
7.1 – Introduction aux taux de réaction
Ce chapitre contient du matériel et des exercices tirés des sections 12.1 “Taux de réaction chimique” et 12.2 “Facteurs affectant les taux de réaction”, et ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
“Taux de réaction chimique,”
Paragraphes 1, 2 et 6 à 9,
Équation 7.1.2, et
“Facteurs affectant les taux de réaction,”
Paragraphes 10 à 14,
Les encadrés “Réaction du césium avec l’eau” et “Facteurs affectant les taux de réaction – activité interactive”, et
Équation 7.1.1.
Ce chapitre contient également des informations tirées de la section 14.1 “Lavitesse d’une réaction chimique” de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0.
Ce chapitre contient des éléments tirés de la section 14.2 – Taux de réaction, y compris les paragraphes 3 à 5 et les équations 7.1.1.
Ce chapitre contient des éléments tirés du paragraphe12.2 – Facteurs affectant les taux de réaction, y compris le paragraphe 15.
Ce chapitre contient des documents originaux de Mahdi Zeghal, notamment le paragraphe 1, la première phrase du paragraphe 9 et la première phrase du paragraphe 12.
Ce chapitre contient la fin du chapitre 7.1, les questions 1 et 2, et la réponse à la question 1 tirée de la section 12.2. “Factors Affecting ReactionRates” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licence CC BY 4.0.
Ce chapitre contient les questions 3 et 4 de la fin du chapitre 7.1, et ses réponses sont tirées de la section 12.1 “Chemical Reaction Rates” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient des réponses originales pour la fin du chapitre 7.1 question 2 créé par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient les figures 7.1.2 et 7.1.3 tirées de “Facteurs affectant les taux de réaction“.
Ce chapitre contient la figure 7.1.1 tirée de 14.2 – Taux de réaction.
7.2 – Mesure et expression des taux de réaction
Ce chapitre contient des éléments tirés de la section 12.1 “Chemical Reaction Rates” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0, notamment :
Les paragraphes 14, 16, 19 et 20, et
Boîte “Taux de réaction dans l’analyse – Bandelettes réactives pour l’analyse d’urine”.
Ce chapitre contient également des informations tirées de la section 14.2 “Mesurer les taux de réaction” de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0, y compris :
Les paragraphes 1 à 13, 24 et 25, et
Équation 7.2.1.
Ce chapitre contient des documents originaux de Mahdi Zeghal, notamment la dernière phrase du paragraphe 3, le paragraphe 11, la dernière phrase du paragraphe 13, le paragraphe 16, 19, 21 et 22.
Ce chapitre contient des documents originaux de Geneviève O’Keefe, y compris le paragraphe 18.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre comprend les figures 7.2.2 et 7.2.5 tirées de “Chemical Reaction Rates“.
7.3 – Lois sur les tarifs
Ce chapitre contient du matériel et des exercices tirés des sections 12.3 “Rate Laws” et 12.4 “Integrated Rate Laws”, et ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
“Lois sur les tarifs“,
Paragraphes 1, 2 et 4-8,
Tableau 7.3.1, et
“Lois sur les tarifs intégrés“
Les paragraphes 17 à 21 et 27, et
Équation 7.3.3.
Ce chapitre contient également du matériel et des extraits des manuels scolaires ouverts suivants :
Sections 12.3 “Rate Laws” et 12.4 “Integrated Rate Laws“, sections de chimie (par la Rice University), utilisées sous une licenceCC BY 4.0,
Sections 14.3 “Concentration et taux (lois de taux différentiels)” et 14.4 “Changement de la concentration avec le temps (lois de taux intégrés)“, sections de la carte de texte Libretextes pour la chimie : The Central Science (par Brown, LeMay, Busten, Murphy et Woodward) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous licenceCC BY-NC-SA 4.0,
Ce chapitre contient le contenu de “Concentration et taux (lois sur les taux différentiels)“, y compris :
Paragraphes 10 à 16,
Exemple 7.3.2,
Équations 7.3.1, et 7.3.2,
“Vérifiez votre apprentissage” 7.3.3,
Tableau 7.3.2
Ce chapitre contient le contenu de “Le changement de concentration avec le temps (lois sur les taux intégrés)“, y compris :
Paragraphes 22-26,
Sections 14.5 “Réactions du premier ordre” et 14.6 “Réactions du second ordre“, sections de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licence CC BY-NC-SA 3.0,
Ce chapitre contient des informations sur les “Réactions de premier ordre“, notamment
Paragraphes 28-30 et 32-36,
Exemple 7.3.5,
“Vérifiez votre apprentissage” 7.3.6,
Ce chapitre contient des éléments tirés de “Réactions de second ordre“, notamment :
Paragraphes 37 à 50,
Équation 7.3.4,
Exemple 7.3.10,
“Vérifiez votre apprentissage” 7.3.11,
Tableau 7.3.3, et
Section 24 “Cinétique“, exercices accompagnant la carte de textes Libretexts de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0.
Ce chapitre comprend des éléments tirés de la section 12.3 – Lois sur les tarifs, y compris :
Paragraphe 2,
Exemples 7.3.1, 7.3.3 et 7.3.6,
“Vérifiez votre apprentissage” 7.3.1, 7.3.4 et 7.3.6.
Ce chapitre comprend des éléments tirés de la section 12.4 – Lois sur les tarifs intégrés, y compris :
Exemples 7.3.7, 7.3.8, 7.3.9 et 7.3.11,
“Vérifiez votre apprentissage” 7.3.8, 7.3.9, 7.3.10 et 7.3.12, et
Paragraphe 51.
Ce chapitre comprend des éléments tirés de l’article 14.3 – Effet de la concentration sur les taux de réaction, notamment :
Exemple 7.3.4, et
“Vérifiez votre apprentissage” 7.3.5.
Ce chapitre contient des documents originaux de Mahdi Zeghal, notamment la quatrième phrase du paragraphe 1 jusqu’à la dernière, la première phrase du paragraphe 19, l’avant-dernière phrase du paragraphe 23 et le paragraphe 31.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des figures, des tableaux et des équations.
Ce chapitre contient les figures 7.3.2, 7.3.3 et 7.3.4 tirées de “Le changement de concentration avec le temps (lois sur les taux intégrés)“.
Ce chapitre contient la figure 7.3.5 tirée de “First-Order Reactions“.
Ce chapitre contient la figure 7.3.8 tirée de “Second-Order Reactions“.
7.4 – Cinétique des réactions : Résumé
Ce chapitre contient des éléments tirés de la section 14.7 “Cinétique des réactions” : A Summary” de la carte de texte des Libretextes de Chimie pour la Chimie Générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0, y compris :
Paragraphes 1 à 4,
Figure 7.4.1,
Exemple 7.4.1, et
“Vérifiez votre apprentissage” 7.4.1.
7.5 – Théorie des collisions
Ce chapitre contient du matériel et des exercices tirés de la section 12.5 “Collision Theory” et ses exercices, respectivement, de la ressource de manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
Les paragraphes 1 à 6, et
Fin du chapitre 7.5 questions et sa réponse 1-8.
Ce chapitre contient des éléments tirés de la section 12.5 – Théorie des collisions, y compris :
Paragraphes 7 à 15,
Exemple 7.5.1, et
“Vérifiez votre apprentissage” 7.5.2.
Ce chapitre contient des réponses originales pour la fin du chapitre 7.5 questions 3 et 5 créées par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres.
Ce chapitre contient la figure 7.5.1 tirée de la “Théorie des collisions“.
Ce chapitre contient les figures 7.5.2, 7.5.3, 7.5.4 et 7.5.5.
7.6 – Théorie de l’État de transition
Ce chapitre contient du matériel tiré des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER) :
Section 9.7 “Theoriesof ReactionRates“, une section de la carte de texte Chemistry Libretexts pour la chimie physique pour les biosciences (par Chang), utilisée sous une licenceCC BY-NC-SA 3.0,
Paragraphes 1-4 et 6-9,
“Gibbs (Free) Energy“, une section de Energies and Potentials (contribution de Doan, Le et Lower) des modules supplémentaires de Chemistry Libretextssur la chimie physique et théorique, utilisée sous licence CC BY-NC-SA 3.0,
La première phrase du paragraphe 5, et
“SN2“, une section de Substitution Reactions (contribution de Curtis, Mooney et Banks) des modules supplémentaires de Chemistry Libretextssur la chimie organique, utilisée sous licenceCC BY-NC-SA 3.0.
Ce chapitre contient des documents originaux de Jessica Thomas, y compris la dernière phrase du paragraphe 5.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres.
Ce chapitre contient la figure 7.6.1 tirée des “Théories des taux de réaction“.
7.7 – Mécanismes de réaction
Ce chapitre contient des éléments tirés de la section 12.6 “Reaction Mechanisms” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient des exercices tirés de la section 14.10 “Mécanismes de réaction” de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0.
Ce chapitre contient des éléments tirés du point 12.6 – Mécanismes de réaction, y compris :
Paragraphes 1-28,
Exemple 7.7.2,
“Vérifiez votre apprentissage” 7.7.2.
Ce chapitre contient des documents originaux de Jessica Thomas, y compris la dernière phrase du paragraphe 25.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres.
Ce chapitre contient la fin du chapitre 7.7 questions 1-3 et ses réponses tirées de 14.10 – Mécanismes de réaction.
Ce chapitre contient les figures 7.7.1 et 7.7.2 tirées de la section 12.6 – Mécanismes de réaction.
7.8 – Catalyse
Ce chapitre contient des éléments tirés de la section 12.7 “Catalyse” du manuel ouvertChemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous une licenceCC BY 4.0.
Ce chapitre contient des éléments tirés du point 12.7 – Catalyse, y compris :
Paragraphes 1, 3-11,
Exemple 7.8.1,
“Vérifiez votre apprentissage” 7.8.1, et
Les boîtes “Mario J. Molina”, “Déficience en glucose-6-phosphate déshydrogénase”, “Convertisseurs catalytiques automobiles” et “Structure et fonction des enzymes”.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris le paragraphe 2 et tous les points directement en dessous et au-dessus du paragraphe 3.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres.
Ce chapitre contient les figures 7.8.1, 7.8.2, 7.8.3, 7.8.4, 7.8.5, 7.8.6 et 7.8.7 tirées du chapitre 12.7 – Catalyse.
Chapitre 7 Termes clés
Les définitions des termes clés suivants ont été adaptées du chapitre 12 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Complexe activé
|
Réaction élémentaire
|
Méthode des taux initiaux
|
Échelon de détermination du taux
|
|
Énergie d’activation (Ea)
|
Catalyseur hétérogène
|
Molécularité
|
Diagramme des coordonnées de réaction
|
|
Équation d’Arrhenius
|
Catalyseur homogène
|
Ordre de réaction global
|
Mécanisme de réaction
|
|
Taux de réaction moyen
|
Taux de réaction initial
|
Facteur pré-exponentiel (A)
|
Ordre de réaction
|
|
Réaction bimoléculaire
|
Taux de réaction instantané
|
Constante de taux (k)
|
Réaction moléculaire
|
|
Catalyseur
|
Loi sur les taux intégrés
|
Loi sur les tarifs
|
Réaction unimoléculaire
|
|
Théorie des collisions
|
Intermédiaire
|
Taux de réaction
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du glossaire du manuel ouvert Introductory Chemistry – 1st Canadian Edition (par Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0 :
|
Complexe activé
|
Catalyseur
|
Catalyseur hétérogène
|
Méthode des taux initiaux
|
|
Énergie d’activation (Ea)
|
Demi-vie d’une réaction (t1/2)
|
Intermédiaire
|
Constante de taux (k)
|
|
|
|
|
La définition du terme clé suivant a été adaptée d’un autre manuel scolaire ouvert du projet LibreTexts de l’Open Education Resource (OER) :
Théorie de l’état de transition – de la section 9.7 “Théories des taux de réaction“, une section de la carte de texte Libretexts pour la chimie physique pour les biosciences (par Chang), utilisée sous une licenceCC BY-NC-SA 3.0.
Fin du chapitre 7 Questions
Ce chapitre contient les questions 1 à 4 traduites et les solutions des examens passés du Dr Alain St-Amant, dont la permission a été accordée.
8 – Structure atomique (théorie quantique et configurations électroniques)
8.1 – La nature de la lumière
Ce chapitre contient des éléments tirés de la section 6.1 “ElectromagneticEnergy” du manuel ouvertChemistry2e (sur OpenStax) de Flowers, Theopold, Langley, and Robinson, PhD, utilisé sous licenceCC BY 4.0, notamment :
Paragraphes 1-9 et 11-19,
Boîte “Communication sans fil” et “Dorothy Hodgkin”,
Exemples 8.1.1, 8.1.2 et 8.1.3,
“Vérifiez votre apprentissage” 8.1.1, 8.1.2 et 8.1.3, et
Équation 8.1.1.
Ce chapitre contient des exercices tirés des ressources de manuels scolaires ouvertes suivantes :
Section 8.1 “Rayonnement électromagnétique“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licence CC BY-NC-SA 3.0,
Fin du chapitre 8.1 questions 8 et 9, et
Section 55 “Light“, une section d’IntroductoryChemistry – 1ère édition canadienne (par Key and Ball), utilisée sous licenceCC BY-NC-SA 4.0,
Fin du chapitre 8.1 questions 1-7.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient un contenu original créé par Jessica Thomas, y compris le paragraphe 10.
Ce chapitre contient les figures 8.1.1, 8.1.2, 8.1.3, 8.1.4, 8.1.5, 8.1.6, 8.1.7, 8.1.8, 8.1.9 et 8.1.10 tirées de “Énergie électromagnétique”.
8.2 – Spectres atomiques
Ce chapitre contient du matériel et des exercices tirés des sections 6.1 “Énergie électromagnétique” et 6.2 “Le modèle de Bohr“, et ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0.
“Énergie électromagnétique“
Paragraphe 1-6,
L’équation 8.2.1, et
“Le modèle Bohr“
Paragraphes 7 à 18,
Équations 8.2.2 et 8.2.3,
Exemples 8.2.1 et 8.2.2, et
“Vérifiez votre apprentissage” 8.2.1 et 8.2.2.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient des réponses originales pour la fin du chapitre 8.2 questions 2 et 8-10 créées par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient la fin des questions du chapitre 8.2 et ses réponses tirées des exercices du chapitre 6.
Ce chapitre contient les figures 8.2.3 et 8.2.4 tirées du “Modèle Bohr“.
Ce chapitre contient les figures 8.2.1 et 8.2.2 extraites de “Énergie électromagnétique“.
8.3 – Dualité matière-énergie des particules d’ondes
Ce chapitre contient des éléments tirés de la section 6.3 “Developmentof Quantum Theory” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0, notamment :
Paragraphes 4 à 12,
Équation 8.3.3 et 8.3.5,
Lien “Dr. Quantum”,
Exemple 8.3.1, et
“Vérifiez votre apprentissage” 8.3.1.
Ce chapitre contient également du matériel/exercices tirés des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER), notamment
“Exercices (problèmes)“, exercices d’un examen du cours CHEM 107B : Physical Chemistry for Life Scientists I (de l’université de Californie Davis), utilisé sous licenceCC BY-NC-SA 3.0,
Fin du chapitre 8.3 questions 3 et 4, et ses réponses,
Section 7.3 “The Wave-Particle Duality of Matter and Energy“, une section de la carte de texte Libretexts pour la chimie – The Molecular Nature of Matter and Change (par Silberberg), utilisée sous une licenceCC BY-NC-SA 3.0,
Paragraphes 1 à 3,
Équations 8.3.1 et 8.3.2,
Fin du chapitre 8.3 questions 1 et 2, et ses exercices, et
Section 8.5 “Deux idées conduisant à une nouvelle mécanique quantique“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.), utilisé sous une licenceCC BY-NC-SA 3.0.
Ce chapitre contient un contenu original créé par Jessica Thomas, notamment la troisième phrase de la dernière phrase du paragraphe 11 et la troisième phrase de la dernière phrase de la légende de la figure 8.3.3.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre comprend les figures 8.3.1 et 8.3.3 tirées de “Developmentof Quantum Theory“.
Ce chapitre comprend les figures 8.3.2 tirées de la section 6.3 – Développement de la théorie quantique.
8.4 – Mécanique quantique
Ce chapitre contient du matériel et des exercices tirés de la section 6.3 “Developmentof Quantum Theory” et de ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0, y compris :
Paragraphes 1-21, et
Équation 8.4.1.
Ce chapitre contient le contenu original du Dr. Kathy Sarah-Focsaneanu, y compris le contenu entre parenthèses de la première phrase du paragraphe 11.
Ce chapitre contient un contenu original créé par Jessica Thomas, notamment la dernière phrase du paragraphe 11 et les trois premières phrases du paragraphe 16.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient la fin du chapitre 8.4 questions 1, 3, 5 et 7 et ses réponses tirées des exercices du chapitre 6.
Ce chapitre contient des réponses originales pour la fin du chapitre 8.4 questions 2, 4, 6 et 8 créées par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient les figures 8.4.1, 8.4.2, 8.4.3, 8.4.4 et 8.4.5 tirées de “Developmentof Quantum Theory“.
8.5 – Configuration des électrons dans les atomes et caractéristiques
Ce chapitre contient du matériel et des exercices tirés des sections 6.3 “Développement de la théorie quantique” et 6.4 “Structure électronique des atomes (configurations électroniques)“, et ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0, y compris :
“Développement de la théorie quantique“,
Paragraphes 1 et 2,
Tableau “Nombres quantiques, leurs propriétés et leur signification”,
Exemples 8.5.1, 8.5.2 et 8.5.3,
“Vérifiez votre apprentissage” 8.5.1, 8.5.2 et 8.5.3, et
“Structure électronique des atomes (configurations électroniques),”
Paragraphe 3-29,
Exemples 8.5.4 et 8.5.5, et
“Vérifiez votre apprentissage” 8.5.4 et 8.5.5.
Ce chapitre contient également des exercices tirés de la section 52 “Organizationof Electronsin Atoms“, une section de l’ouvrage “IntroductoryChemistry – 1st Canadian Edition” (par Key and Ball), utilisé sous licenceCC BY-NC-SA 4.0.
Ce chapitre contient le contenu original du Dr. Kathy Sarah-Focsaneanu, y compris la figure 8.5.1, tirée de son contenu de cours.
Ce chapitre contient le contenu original créé par Jessica Thomas, y compris les quatre dernières phrases de la légende de la figure 8.5.3.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient la fin du chapitre 8.5, les questions de 1 à 9 et ses réponses sont tirées de la section 6.4 “ElectronicStructure of Atoms(ElectronConfigurations)” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient la fin du chapitre 8.5 des questions de 10-17 et ses réponses tirées de la section 52 “Organisation des électrons dans les atomes” de l’ouvrage Introductory Chemistry – 1st Canadian Edition (by Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0.
Ce chapitre contient les figures 8.5.1, 8.5.2, 8.5.3, 8.5.4 et 8.5.5 tirées de la section 6.4 “ElectronicStructure of Atoms(ElectronConfigurations)” du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient la figure 8.5.7 tirée de la page Chromium de WebElements, https://www.webelements.com, consultée en août 2020.
8.6 – Propriétés atomiques générales
Ce chapitre contient des exercices tirés du chapitre 8 Exercices du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Ce chapitre contient également du matériel et/ou des exercices tirés des manuels scolaires ouverts suivants du projet LibreTexts Open Education Resource (OER), notamment
Section 4.1 “Ions – Perte et gain d’électrons“, une unité du cours CHE 1305 – Introductory Chemistry (de la Lubbock Christian University), utilisée sous licence CC BY-NC-SA 3.0,
Le paragraphe 1, et
Section 9.6 “Propriétés magnétiques“, une section de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.), utilisé sous une licenceCC BY-NC-SA 3.0,
Paragraphes 2 à 8,
“Note” sous les paragraphes 4 et 6,
Exemples 8.6.1 et 8.6.2,
“Vérifiez votre apprentissage” 8.6.1 et 8.6.2.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres.
Ce chapitre contient la fin du chapitre 8.6, questions 1 et 2, et ses réponses sont tirées de _COPY4@12.1:M3s_Xl4y@12/Molecular-Orbital-Theory
Ce chapitre contient la fin du chapitre 8.6 questions 3-5, et ses réponses tirées de “Propriétés magnétiques“.
Ce chapitre contient la figure 8.6.1 tirée de “Ions – Losing and Gaining Electrons“.
Ce chapitre contient les figures 8.6.2 tirées de “Propriétés magnétiques“.
8.7 – Tendances périodiques et variation des propriétés
Ce chapitre contient du matériel et des exercices tirés de la section 6.5 “PeriodicVariations in Element Properties” et ses exercices, respectivement, de la ressource de manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
Paragraphes 1 à 22,
Exemples 8.7.1 et 8.7.2,
“Vérifiez votre apprentissage” 8.7.1 et 8.7.2, et
Tableaux 8.7.1 et 8.7.2.
Ce chapitre contient également du matériel et des exercices tirés de la section 8.4 “Bond Polarity and Electronegativity” de la carte de texte Libretexts pour la chimie : The Central Science (par Brown, LeMay, Busten, Murphy et Woodward) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous licenceCC BY-NC-SA 4.0, y compris :
Paragraphes 23 à 30,
Ce chapitre contient des documents originaux de Jessica Thomas, y compris la légende de la figure 8.7.10.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des figures et des tableaux.
Ce chapitre contient les figures 8.7.1, 8.7.2, 8.7.3, 8.7.5, 8.7.6 et 8.7.7 tirées de “PeriodicVariations in Element Properties“.
Ce chapitre contient les figures 8.7.8, 8.7.9 et 8.7.10 tirées de “Bond Polarity and Electronegativity“.
Ce chapitre contient la figure 8.7.4 tirée de la section 3.4 “Quantification de l’énergie et configuration des électrons” du manuel de référence ouvert Siyavula : Sciences physiques de 10e année (sur OpenStax) par le Free High School Science Texts Project, utilisé sous une licenceCC BY 3.0.
Chapitre 8 Termes clés
La définition du terme clé “amplitude” a été adaptée de la section 6.1 “Énergie électromagnétique” du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0.
Les définitions des termes clés suivants ont été adaptées du chapitre 6 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Nombre quantique du moment angulaire(ℓ)
|
Spectre électromagnétique
|
Isoélectronique
|
orbitale s
|
|
Orbite atomique
|
Affinité électronique (EA)
|
Spectre de lignes
|
Shell
|
|
Le principe d’Aufbau
|
Configuration des électrons
|
Nombre quantique magnétique(mℓ)
|
Nombre de spins quantiques (ms)
|
|
Corps noir
|
Un état électronique excité
|
Nœud
|
Vague permanente
|
|
Le modèle de Bohr
|
orbitale f
|
Diagramme orbital
|
Sous-coque
|
|
Spectre continu
|
Fréquence(ν)
|
orbitale p
|
Électrons de valence
|
|
Noyau électronique
|
État électronique de base
|
Le principe d’exclusion de Pauli
|
Coquille de Valence
|
|
Rayon de covalence
|
Le principe d’incertitude de Heisenberg
|
Photon
|
Vague
|
|
orbitale d
|
Hertz (Hz)
|
Nombre quantique principal (n)
|
La dualité onde-particule
|
|
Orbites dégénérées
|
La règle de Hund
|
Quantification
|
Wavefunction(ѱ)
|
|
Charge nucléaire effective (Zeff)
|
Modèle d’interférence
|
Mécanique quantique
|
Longueur d’onde(λ)
|
|
Rayonnement électromagnétique
|
Énergie d’ionisation (IE)
|
Nombre quantique
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du glossaire du manuel ouvert Introductory Chemistry – 1st Canadian Edition (par Key and Ball), utilisé sous une licenceCC BY-NC-SA 4.0 :
|
Orbite atomique
|
Spectre continu
|
Spectre électromagnétique
|
Électrons de valence
|
|
Le principe d’Aufbau
|
Charge nucléaire effective (Zeff)
|
Spectre de lignes
|
Coquille de Valence
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées à partir d’autres ressources de manuels scolaires ouverts du projet LibreTexts de ressources éducatives ouvertes (REL) :
Diamagnétisme, moment magnétique et paramagnétisme – de la section 9.6 “Propriétés magnétiques“, une section de la carte de textes Libretexts de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.), utilisé sous licenceCC BY-NC-SA 3.0, et
Blindage – de la section 6.17 “Blindage électronique”, une section de l’introduction à la chimie (CK-12), utilisée sous une licenceCC BY-NC 4.0.
Fin du chapitre 8 Questions
Ce chapitre contient les questions 1-2 traduites et les solutions des examens passés du Dr Alain St-Amant, dont la permission a été accordée.
9 – Liaison moléculaire
9.1 – Propriétés atomiques des liaisons chimiques
Ce chapitre contient du matériel et des exercices tirés de la section 7.3 “Lewis Symbols and Structures” et ses exercices, respectivement, de la ressource de manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0.
Ce chapitre contient également des éléments tirés de la section 10.1 “Théorie de Lewis” : An Overview” de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
9.2 – Liaison ionique
Ce chapitre contient du matériel et des exercices tirés des sections 7.1 “Liaison ionique” et 7.5 “Résistance des liaisons ioniques et covalentes“, et ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0, y compris :
Ce chapitre contient des informations sur les “liaisons ioniques“, notamment les paragraphes 1 à 4 et la première phrase du paragraphe 5,
Ce chapitre contient du matériel tiré de “Strengthsof Ionic and Covalent Bonds”, y compris les paragraphes 6 à 8, l’exemple 9.2.1 et “Check your learning” 9.2.1.
Ce chapitre contient le contenu original du Dr. Kathy Sarah-Focsaneanu, y compris la dernière phrase des paragraphes 4 et 5.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres et des équations.
Ce chapitre contient la fin du chapitre 9.2 questions et réponses pour les exercices 1 à 9 tirés du chapitre 7.
Ce chapitre contient les figures 9.2.1 et 9.2.1 tirées de “Ionic Bonding“.
9.3 – Liaisons covalente
Ce chapitre contient du matériel et des exercices tirés des sections 7.2 “Covalent Bonding” et 7.5 “Strengthsof Ionic and Covalent Bonds“, et ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
“Liaison covalente“, y compris les paragraphes 1 à 16, les exemples 9.3.1, “Vérifiez votre apprentissage” 9.3.1 et le tableau 9.3.1, et
“Strengths of Ionic and Covalent Bonds“, y compris les paragraphes 17-22 et le tableau 9.3.2.
Ce chapitre contient le contenu original du Dr. Kathy Sarah-Focsaneanu, y compris la dernière phrase de la légende de la figure 9.3.1 et la dernière phrase du paragraphe 13.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris la deuxième phrase du paragraphe 15.
Ce chapitre contient les réponses originales aux questions 1 et 7 créées par Nathan Biniam et Leanne Trepanier.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des figures et des tableaux.
Ce chapitre comprend les figures 9.3.1, 9.3.2, 9.3.3 et 9.3.4 tirées de “Covalent Bonding“.
9.4 – Représentation des molécules et des ions avec des structures de Lewis
Ce chapitre contient du matériel et des exercices tirés des sections 7.3 “Symboles et structures de Lewis” et 7.4 “Charges formelles et résonance“, et ses exercices, respectivement, du manuel ouvert Chemistry2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0, y compris :
“Symboles et structures de Lewis,”
Les paragraphes 1 à 12, 14 à 25 et tous les points soulevés dans le cadre de ces paragraphes,
Exemples 9.4.1, et 9.4.3,
“Vérifiez votre apprentissage” 9.4.1, 9.4.3,
“Charges formelles et résonance,”
Les paragraphes 25 à 35, 37 à 40 et tous les points soulevés dans le cadre de ces paragraphes,
Exemples 9.4.4, et 9.4.5 et 9.4.6,
“Vérifiez votre apprentissage” 9.4.4, et 9.4.5 et 9.4.6, et
Chapitre 7 Exercices,
La fin du chapitre 9.4 question 1-8 et ses réponses.
Ce chapitre contient également des éléments tirés de la section 10.2 “Liaisons covalentes” : Introduction” de la carte de texte des Libretextes de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licence CC BY-NC-SA 3.0.
Ce chapitre contient du matériel de la section 8.5 – Dessin des structures de Lewis, vu dans l’exemple 9.4.2 et “Vérifiez votre apprentissage” 9.4.2.
Ce chapitre contient le contenu original du Dr. Kathy Sarah-Focsaneanu, y compris la partie entre parenthèses dans la deuxième phrase du paragraphe 23.
Ce chapitre contient le contenu original de Mahdi Zeghal, y compris le 13e paragraphe et la dernière phrase des paragraphes 36 et 40.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres.
Ce chapitre contient la figure 9.4.1 tirée de “Lewis Symbols and Structures“.
9.5 – VSEPR
Ce chapitre contient du matériel et des exercices tirés de la section 7.6 “Structure moléculaire et polarité” et ses exercices, respectivement, de la ressource de manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0, y compris :
Paragraphes 1-28, et les points numérotés du paragraphe 15,
Exemples 9.5.1, 9.5.2, 9.5.3, 9.5.4, 9.5.5 et 9.5.7,
“Vérifiez votre apprentissage” 9.5.1, 9.5.2, 9.5.3, 9.5.4, 9.5.5, et 9.5.7, et
Activité interactive “VSEPR” et “Polarité moléculaire”.
Ce chapitre contient également des informations tirées de la section 10.7 “Formes des molécules” de la carte de texte Libretexts de chimie pour la chimie générale : Principles and Modern Applications (par Petrucci et al.) dans le cadre du projet LibreTexts Open Education Resource (OER), utilisé sous une licenceCC BY-NC-SA 3.0, y compris :
Exemple 9.5.6, et
“Vérifiez votre apprentissage” 9.5.6.
Ce chapitre contient le contenu original de Geneviève O’Keefe et Derek Fraser-Halberg, y compris la numérotation des chiffres.
Ce chapitre contient le contenu original de Derek Fraser-Halberg, y compris la partie de la légende entre parenthèses du point a) de la figure 9.5.10.
Ce chapitre contient des questions de fin de chapitre 9.5 de 1 à 12 et ses réponses sont tirées des exercices du chapitre 7.
Ce chapitre contient la figure 9.5.7 tirée de la section 10.2 – Théorie de la VSEPR – Les cinq formes de base.
Ce chapitre contient les figures 9.5.1, 9.5.2, 9.5.3, 9.5.4, 9.5.5, 9.5.6, 9.5.8, 9.5.9, 9.5.10, 9.5.11, 9.5.12, 9.5.13, 9.5.14, et 9.5.15 tirées de “Structure moléculaire et polarité“.
Chapitre 9 Termes clés
Les définitions des termes clés suivants ont été adaptées du chapitre 7 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Position axiale
|
Position équatoriale
|
Structure moléculaire
|
Lien simple
|
|
Angle de liaison
|
Octuor élargi
|
Octahedral
|
Tétraèdre
|
|
Moment de dipôle de liaison
|
Charge formelle
|
La règle de l’octuor
|
Trigonal bipyramidal
|
|
Énergie de dissociation des obligations (BE)
|
Radical libre
|
Liaison covalente polaire
|
Trigonal planar
|
|
Longueur de la caution
|
Énergie en treillis(ΔHlattice)
|
Molécule polaire
|
Triple obligation
|
|
Moment de dipôle
|
Structure de Lewis
|
Liaison covalente pure
|
Théorie de la répulsion des paires d’électrons de la coque de Valence (VSEPR)
|
|
Double lien
|
Le symbole de Lewis
|
Résonance
|
Vecteur
|
|
Géométrie des paires d’électrons
|
Linéaire
|
Formes de résonance
|
|
|
Electronégativité (EN)
|
Paire d’appareils
|
Hybride de résonance
|
|
|
|
|
|
Les définitions des termes clés suivants ont été adaptées du chapitre 8 Termes clés du manuel ouvert Chemistry2e (sur OpenStax) par Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous une licenceCC BY 4.0 :
|
Ordonnance de cautionnement
|
Liaison covalente polaire
|
|
|
|
|
|
|
Fin du chapitre 9 Questions
Ce chapitre contient les questions 1-3 traduites et les solutions des examens passés du Dr. Alain St-Amant, dont la permission a été accordée.
Annexes
Pour les annexes non incluses ici, leurs données/contenu ont été extraits de sources non-OER (par ex. journal, base de données en ligne) et celles-ci ont été référencées à la fin dans une section “Références”.
Annexe C | Mathématiques essentielles
Cette annexe contient des éléments tirés de l’annexe B “Essential Mathematics” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous une licenceCC BY 4.0. Les modifications/ajouts au matériel ont été faits par Mahdi Zeghal, comme indiqué ci-dessous.
Cet appendice contient le contenu original de Mahdi Zeghal, y compris le remplacement de l’expression “arithmétique exponentielle” par “notation scientifique” ou “notation exponentielle”, le paragraphe et l’exemple sur les opérations logarithmiques avec des chiffres significatifs dans la sous-section “Chiffres significatifs”,la note se référant à la section “Chiffres significatifs et incertitude” du chapitre d’introduction qui détaille les chiffres significatifs, le premier paragraphe, la première phrase du deuxième paragraphe, et le troisième paragraphe et la dérivation algébrique de la formule quadratique dans la sous-section “La solution des équations quadratiques”, et les deux graphiques de la sous-section “Graphique bidimensionnel (x – y)”.”
Annexe D | Unités et facteurs de conversion
Cette annexe contient des informations tirées de l’annexe C “Unités et facteurs de conversion” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0. Cela comprend les tableaux D.2 à D.5 et le tableau D.7.
Cette annexe contient également des éléments tirés du chapitre 3 “Multiples et sous-multiples décimaux des unités SI” de la brochure Le Système international d’unités (SI) du Bureau international des poids et mesures (BIPM), utilisée sous licenceCC BY 4.0. Cela inclut le tableau D.1.
Cet appendice contient également le contenu original de Mahdi Zeghal, y compris l’inclusion de l’unité SI pour chaque variable en haut des tableaux D.2 à D.7, la définition d’une livre en onces, la définition d’une unité de masse atomique (amu), les définitions d’un joule, le tableau “Unités de température”, et la définition d’une atmosphère (atm) en N m-2.
Cette annexe contient également du matériel tiré de “Dimensional Analysis” de la ressource pédagogique ouverte et du cours Boundless Chemistry (sur Lumen Learning), utilisé sous une licenceCC BY 4.0. Cela inclut tout le matériel sous la rubrique “Analyse dimensionnelle” (sauf la deuxième phrase du premier paragraphe et le diagramme visuel de l’équation), qui a été réorganisé et organisé différemment dans certains paragraphes (en particulier, les paragraphes deux, quatre et cinq.
Cette annexe contient également des éléments tirés des notes de cours du Dr Kathy-Sarah Focsaneanu, y compris la deuxième phrase et le diagramme visuel de l’équation sous la rubrique “Analyse dimensionnelle”.
Annexe E | Formules et constantes physiques fondamentales
Cette annexe contient du matériel tiré de la fiche de données du cours du Dr Kathy-Sarah Focsaneanu, y compris tout le matériel sous la rubrique “Formules clés”.
Annexe F | Propriétés de l’eau
Cette annexe contient des informations tirées de l’annexe E “Water Properties” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0. Tous les chiffres sont inclus.
Cette annexe contient également des informations tirées de la fiche de données du cours du Dr Kathy-Sarah Focsaneanu, y compris le tableau F.4.
Annexe G | Enthalpies standard de formation pour certaines substances
Cette annexe contient des informations tirées de l’annexe G “Standard Thermodynamic Properties for Selected Substances” du manuel ouvertChemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0. Cela inclut les données pour toutes les substances qui ne sont pas trouvées dans la référence de l’annexe.
Annexe J | Constantes de formation pour les ions complexes
Cette annexe contient des informations tirées de l’annexe G “Formation Constants for Complex Ions” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley, et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Annexe K | Règles de solubilité pour les composés ioniques communs dans l’eau
Cette annexe contient des informations tirées de la section 4.2 “ClassifyingChemical Reactions” du manuel ouvert Chemistry (sur BC Open Textbooks) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous licenceCC BY 4.0.
Annexe L | Produits de solubilité des sels communs
Cette annexe contient des informations tirées de l’annexe J “Solubility Products” du manuel ouvert Chemistry 2e (sur OpenStax) de Flowers, Theopold, Langley et Robinson, PhD, utilisé sous une licenceCC BY 4.0. Cela inclut les données pour toutes les substances qui ne sont pas trouvées dans la référence de l’annexe.


















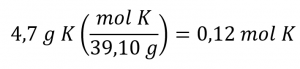





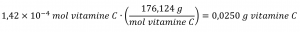
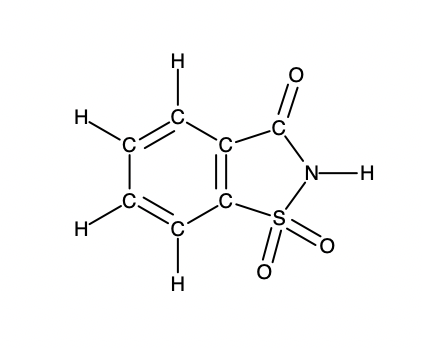
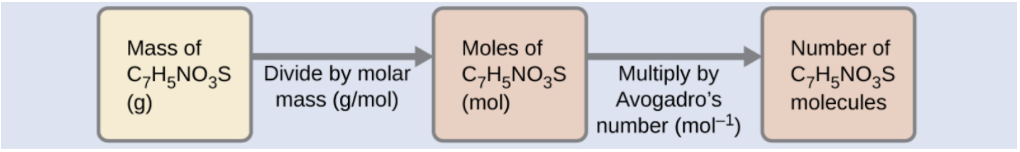
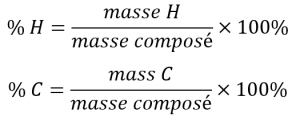
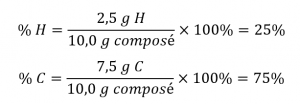
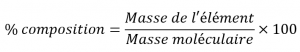
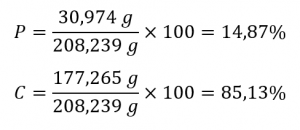
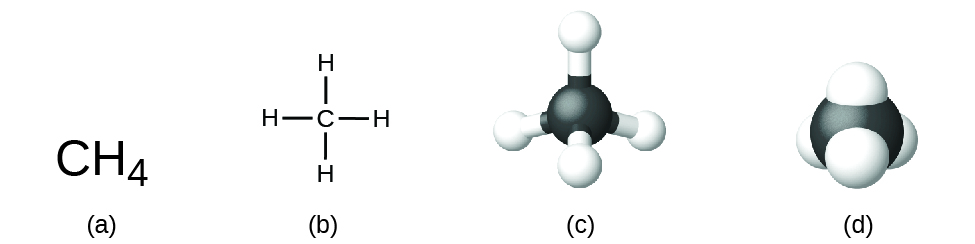
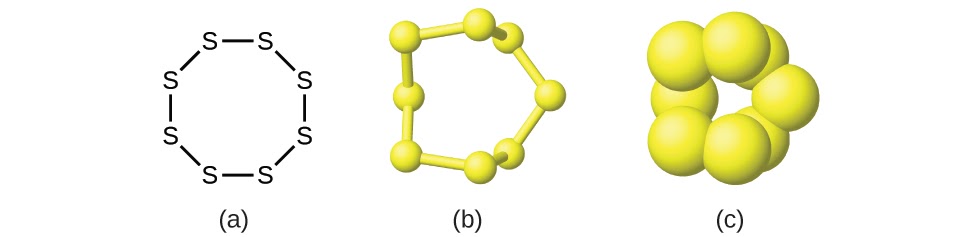
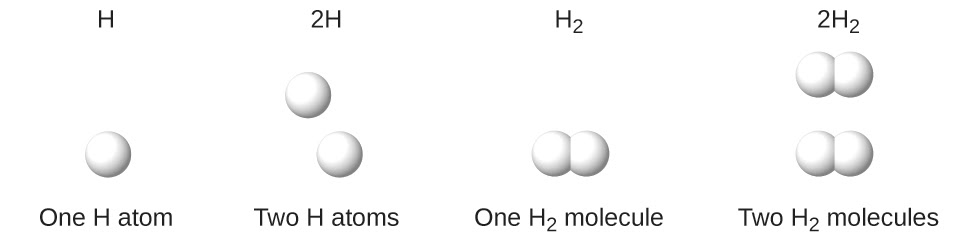
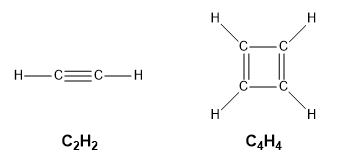
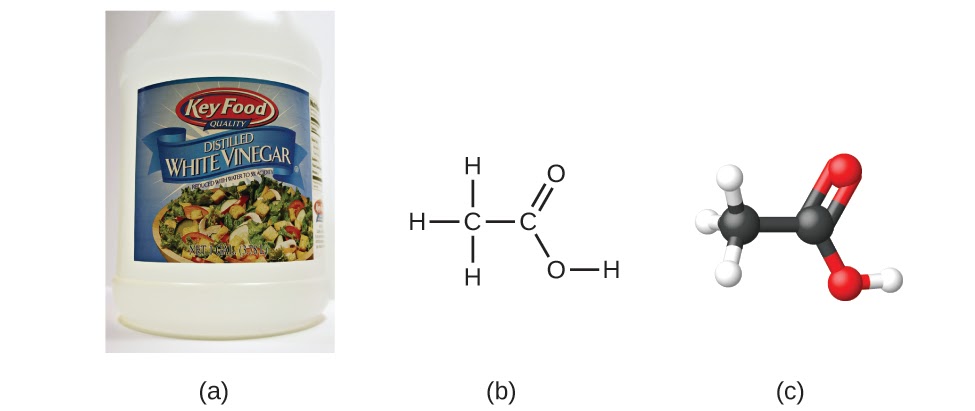
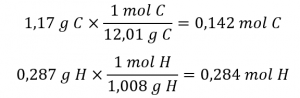
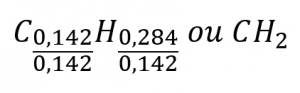
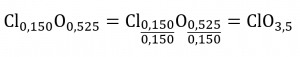
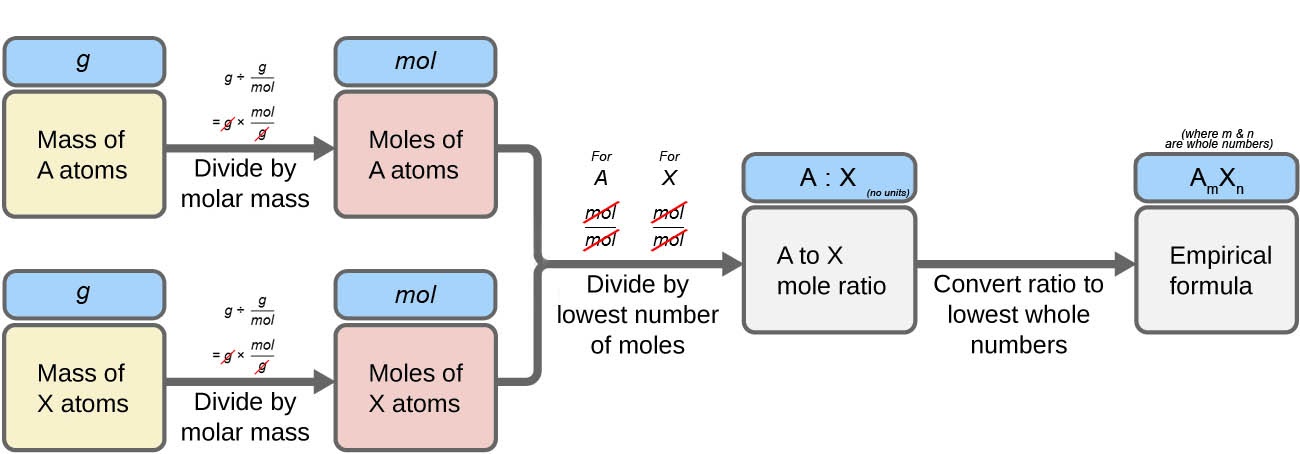

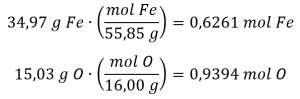
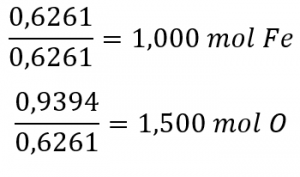

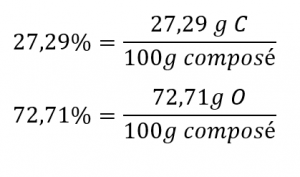
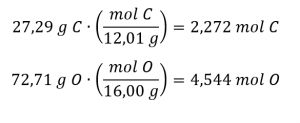
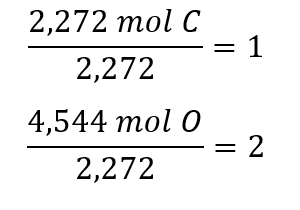
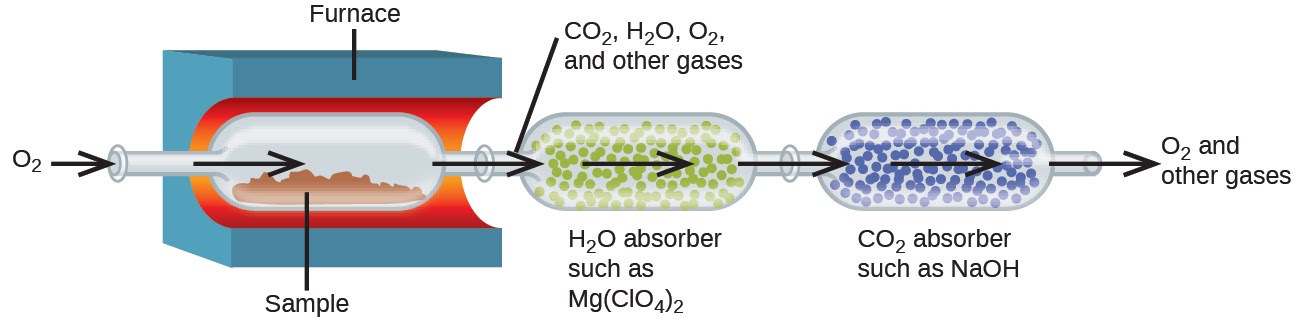
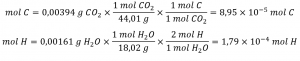
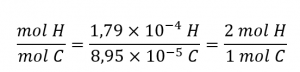
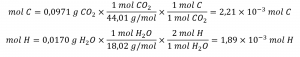
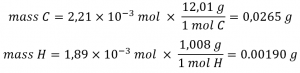
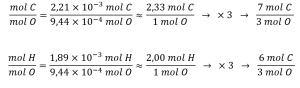
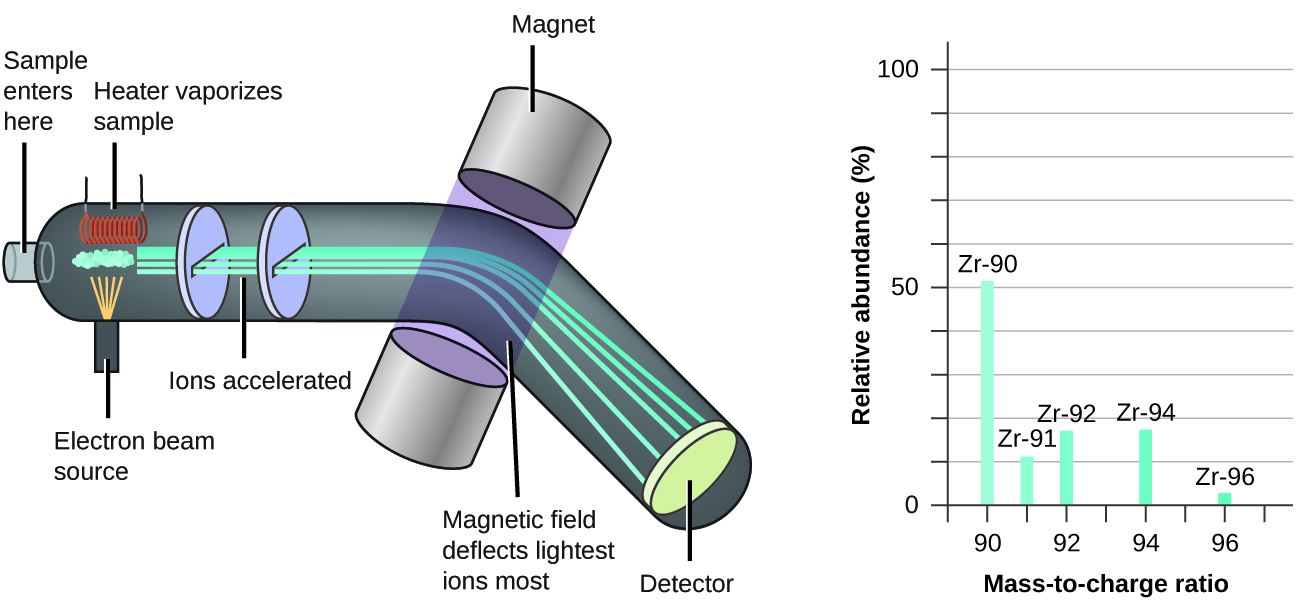
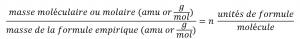
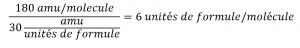
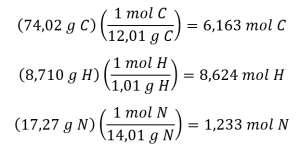

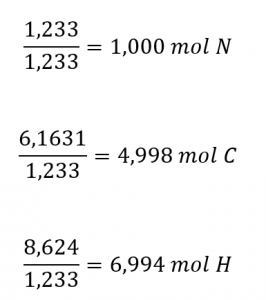 Les rapports molaires C-N et H-N sont suffisamment proches des nombres entiers, et la formule empirique est donc
Les rapports molaires C-N et H-N sont suffisamment proches des nombres entiers, et la formule empirique est donc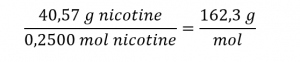
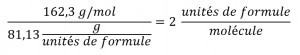
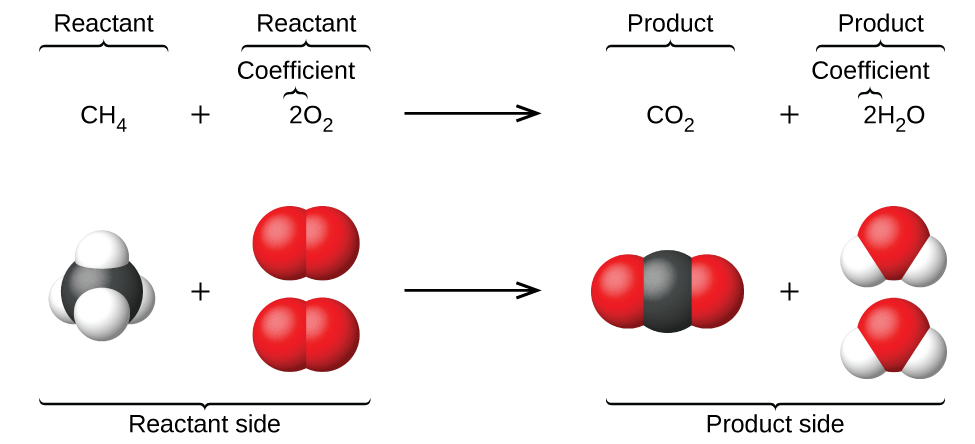
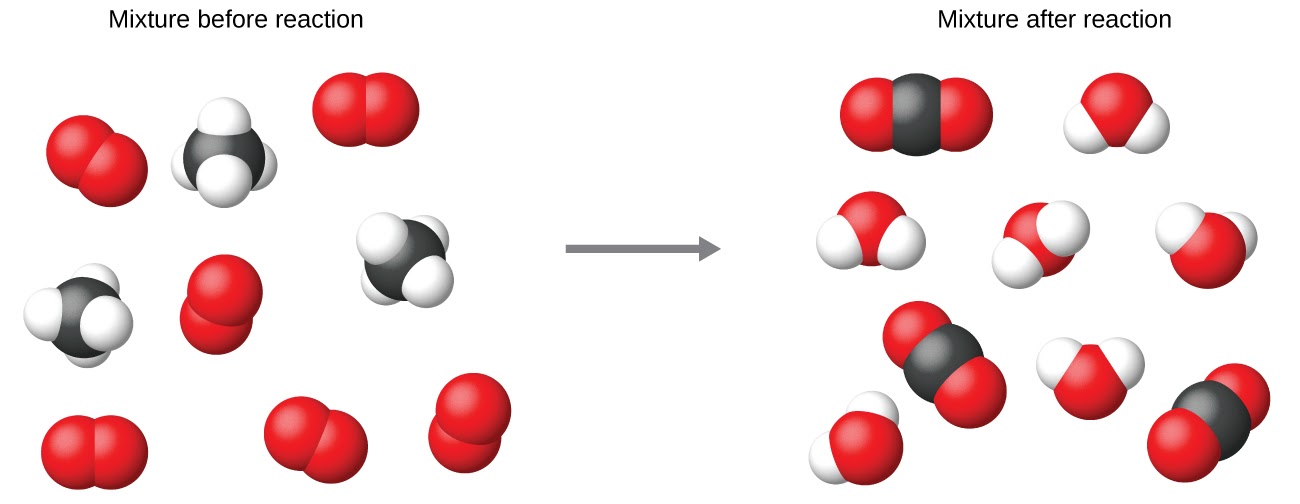
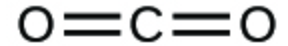

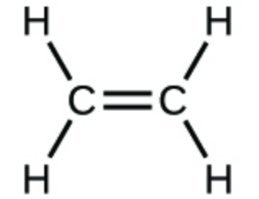
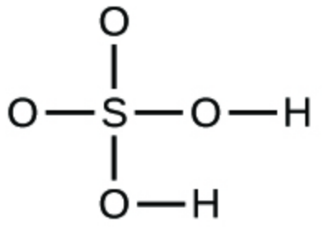
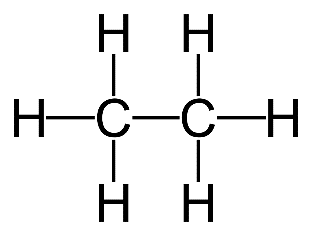
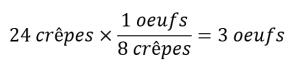


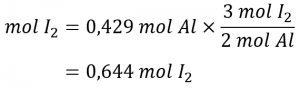

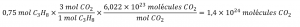
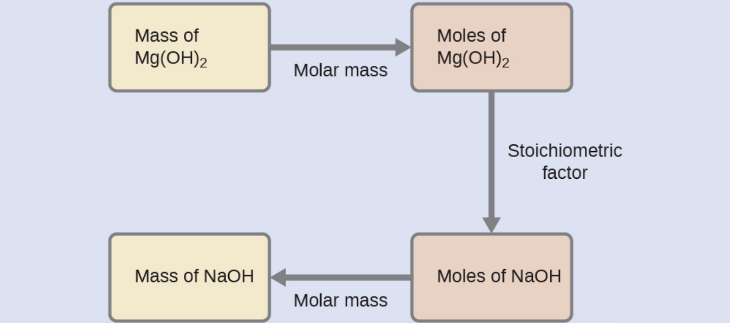
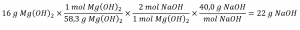
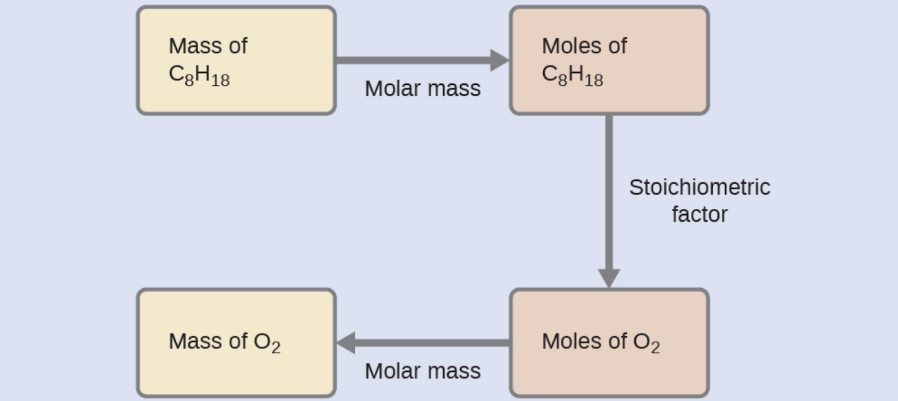
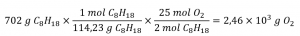
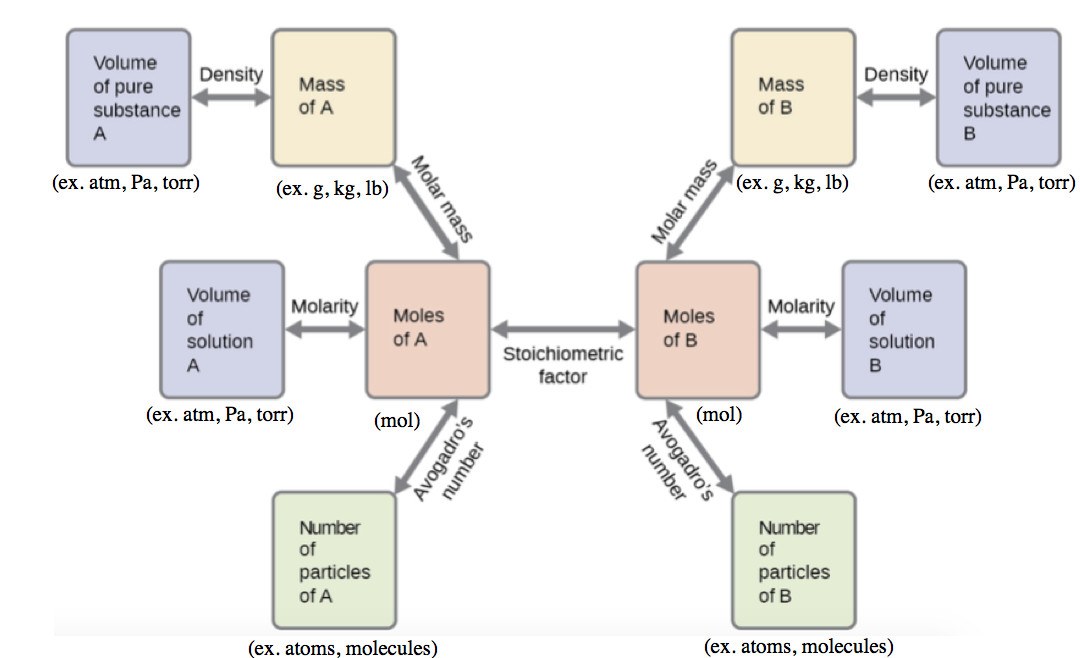
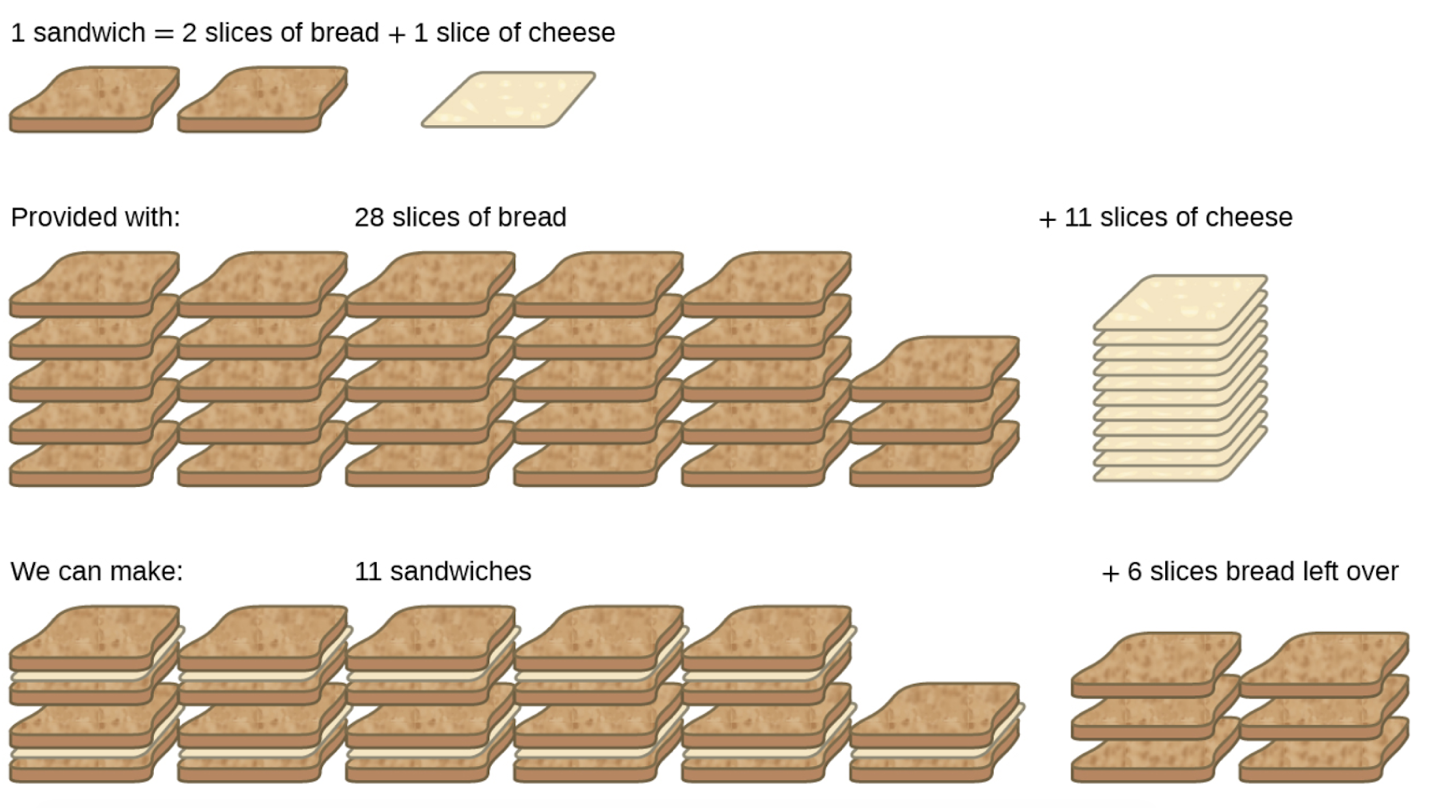
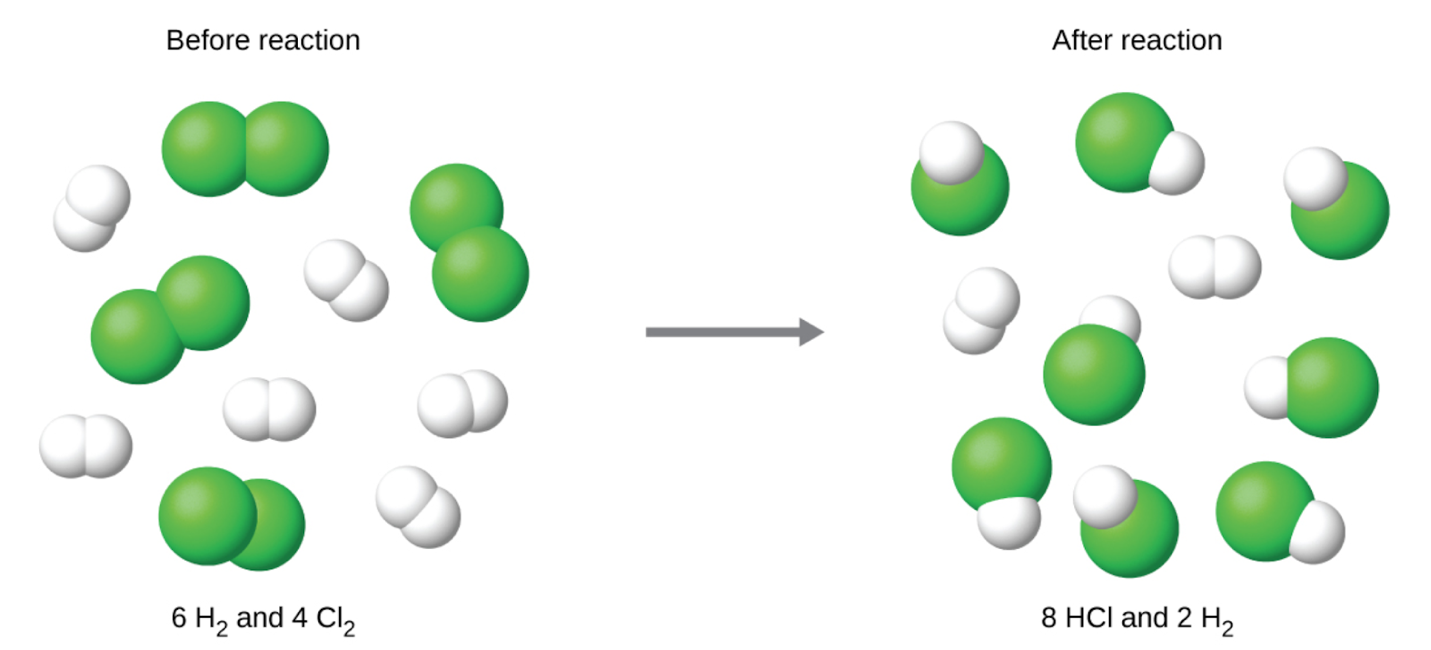
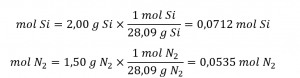
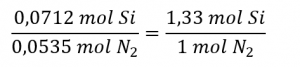
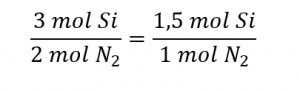
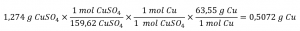


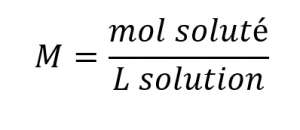
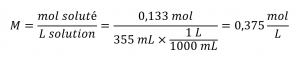

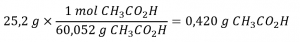
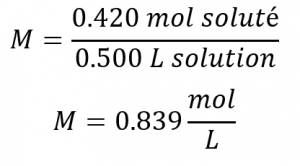
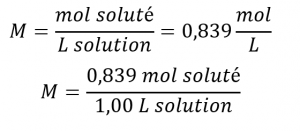
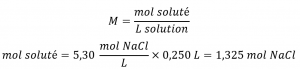
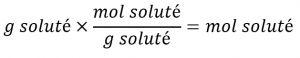
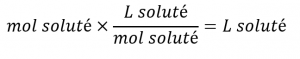
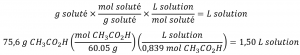

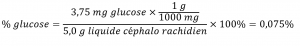
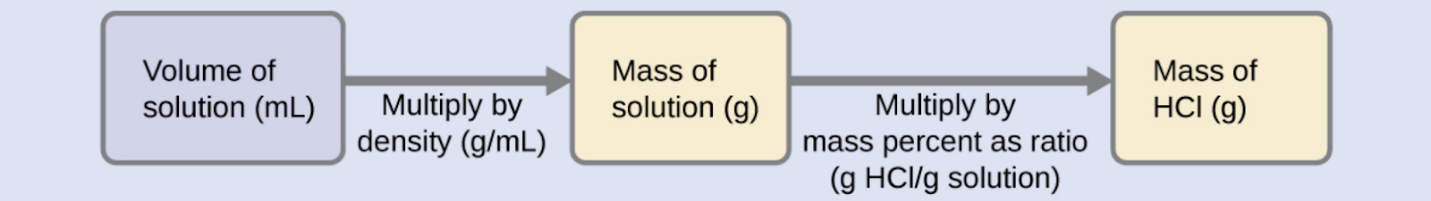
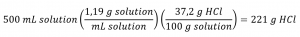


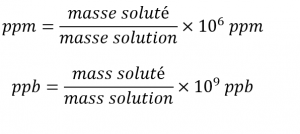

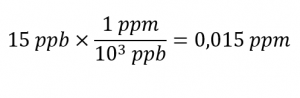

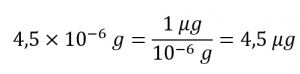
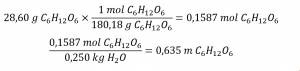


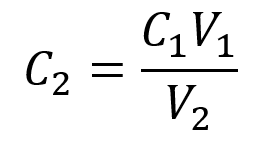
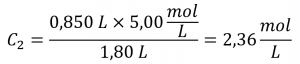
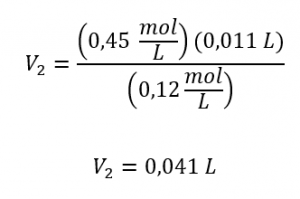
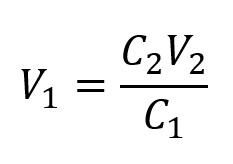
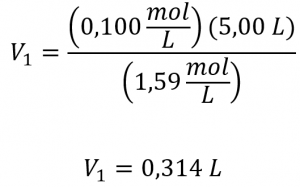
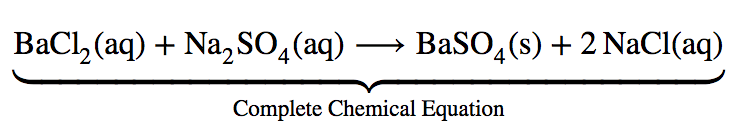
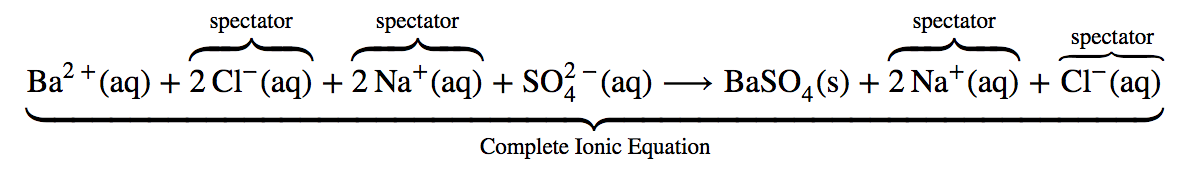
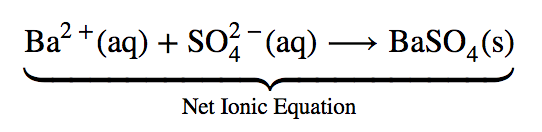



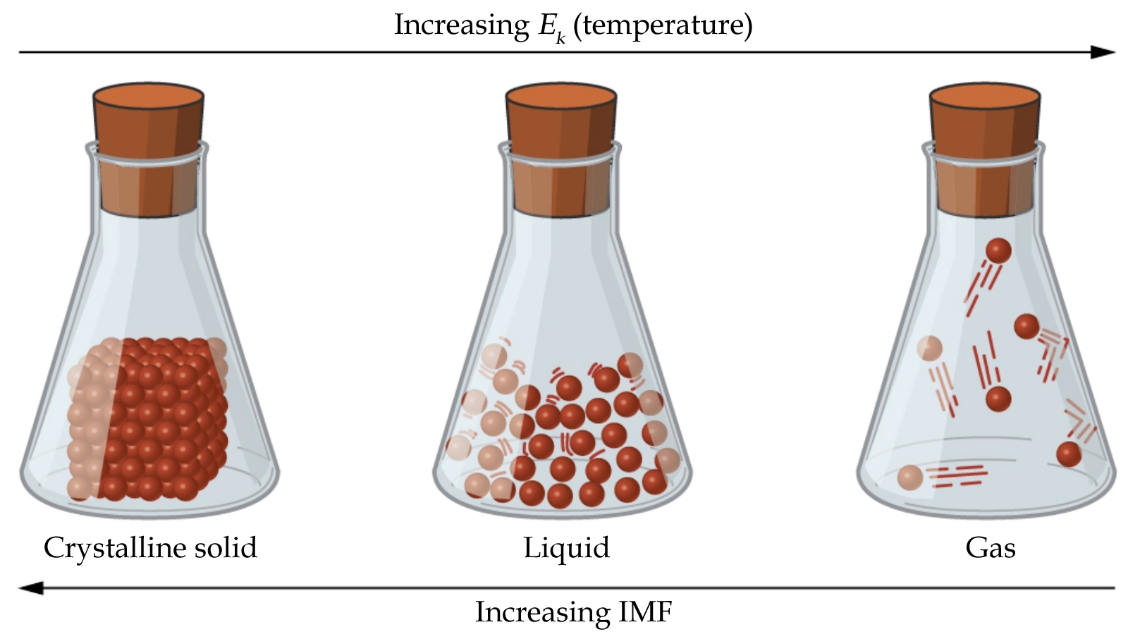


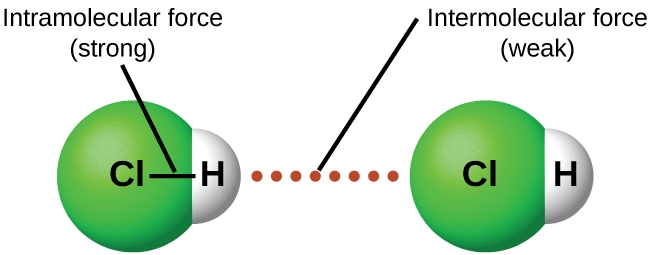
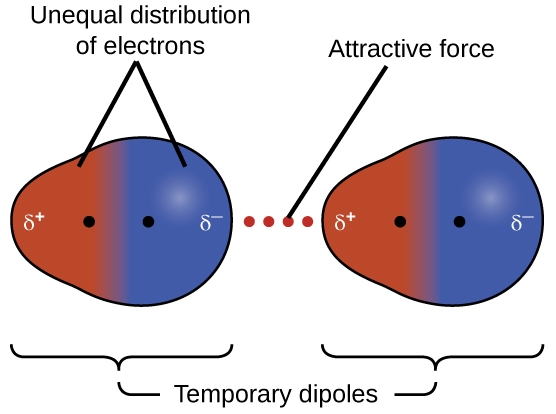
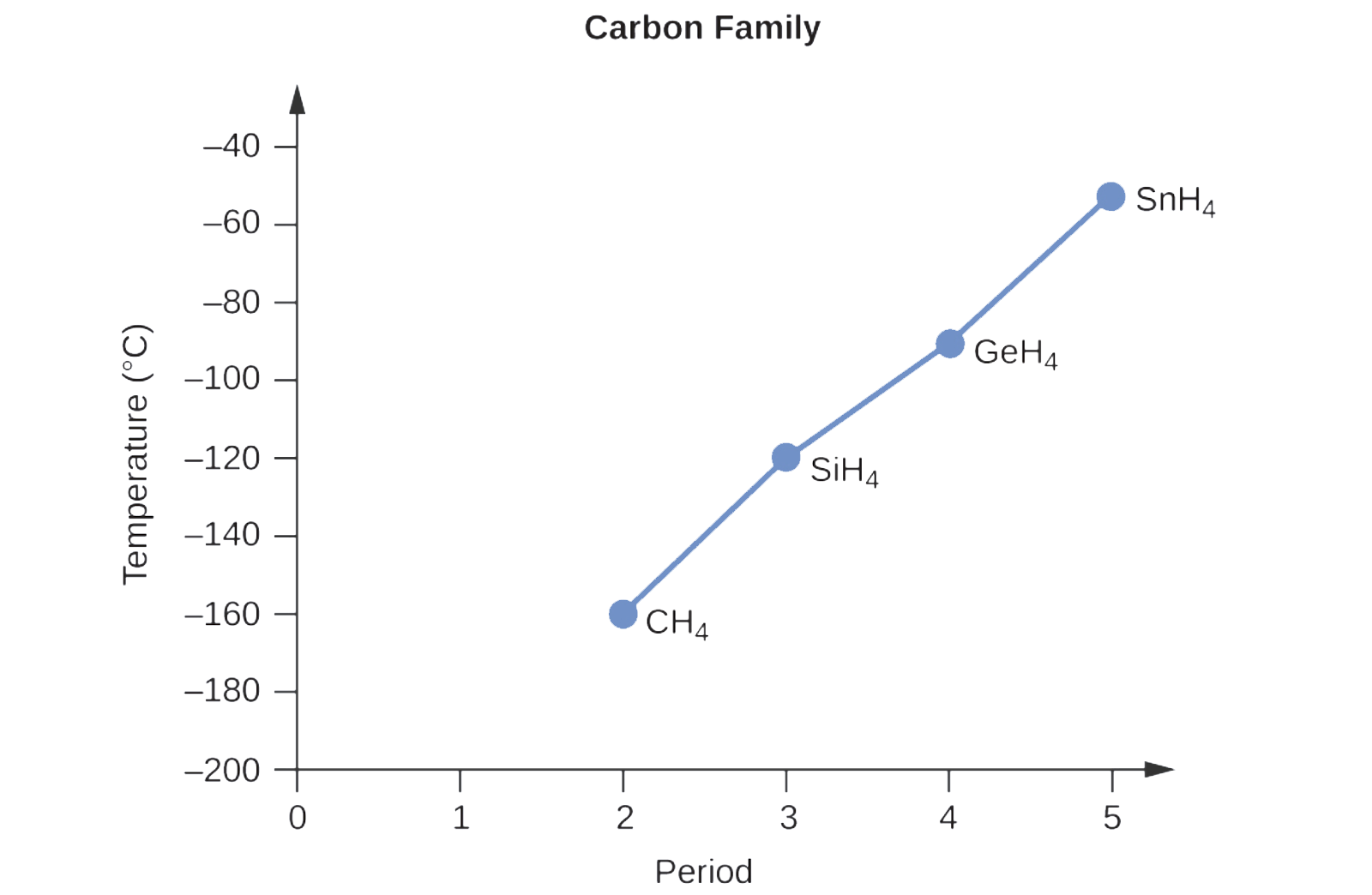
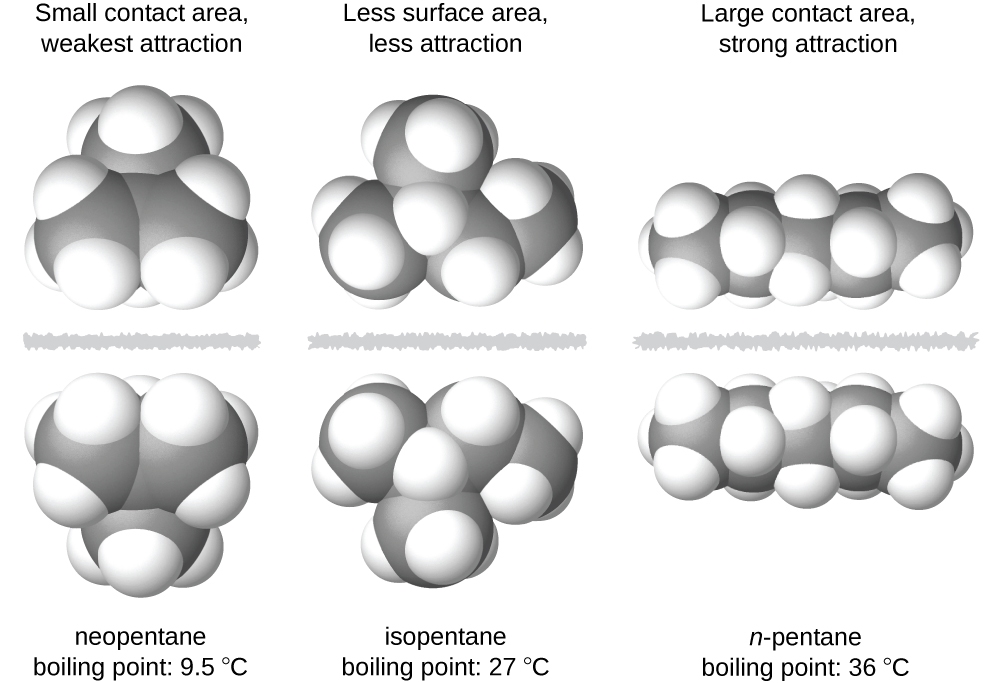
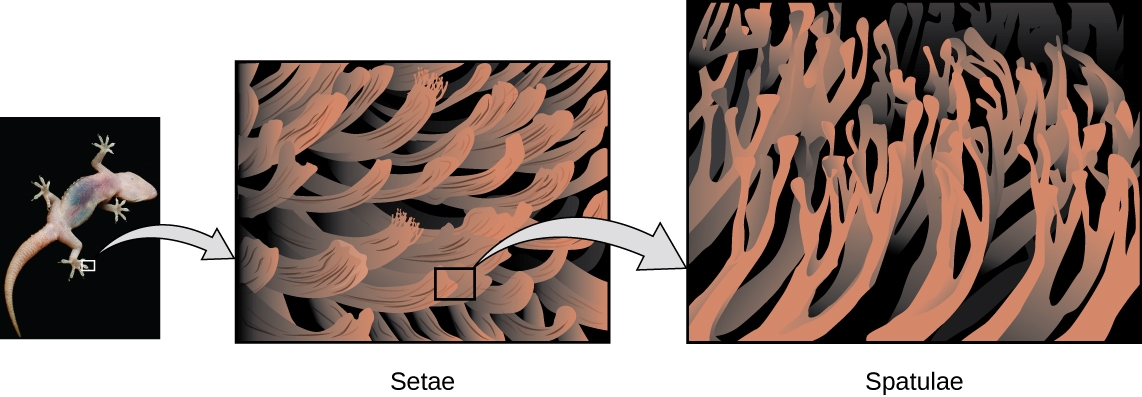
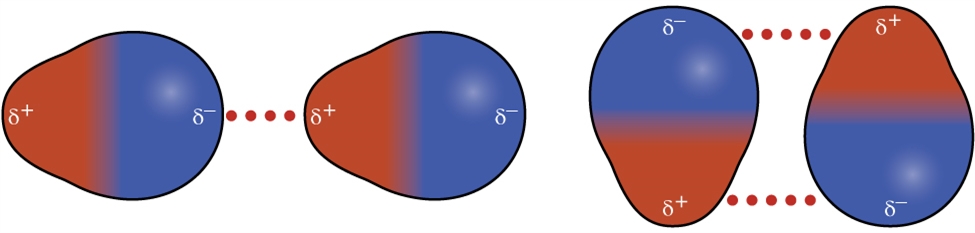
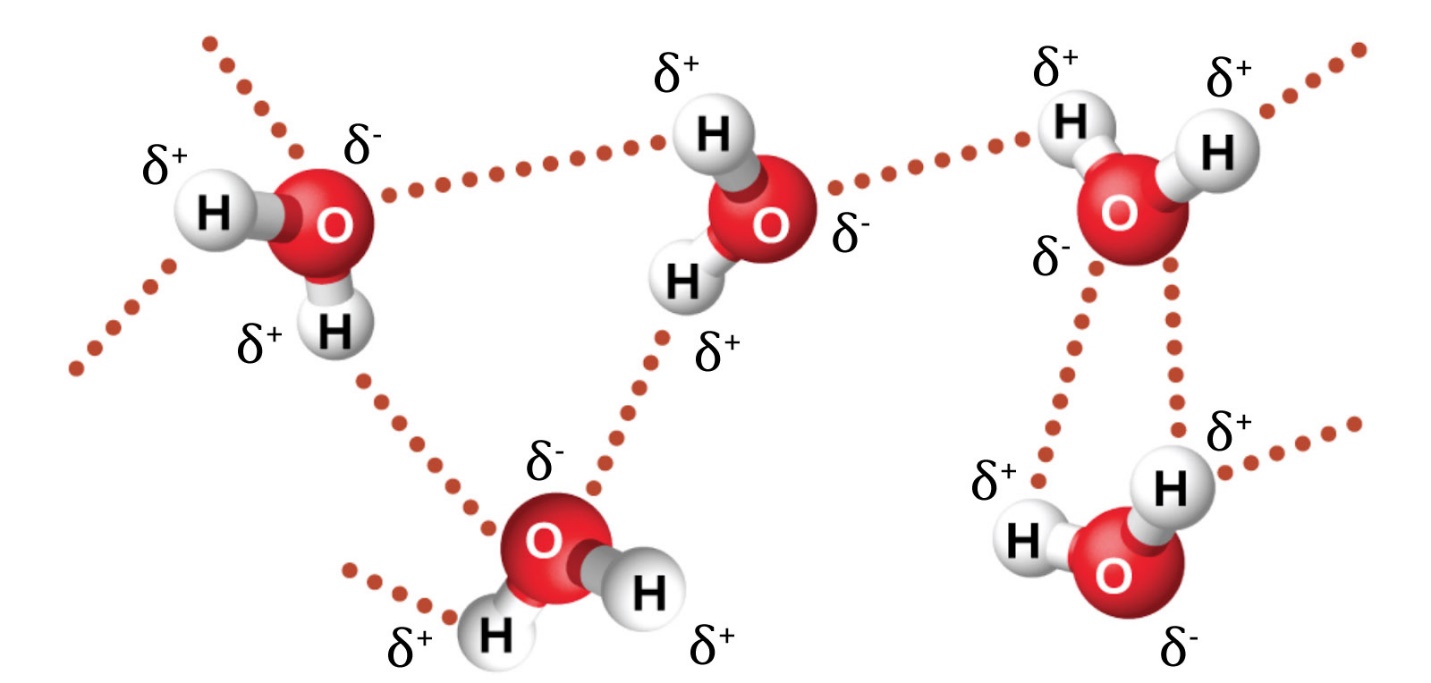
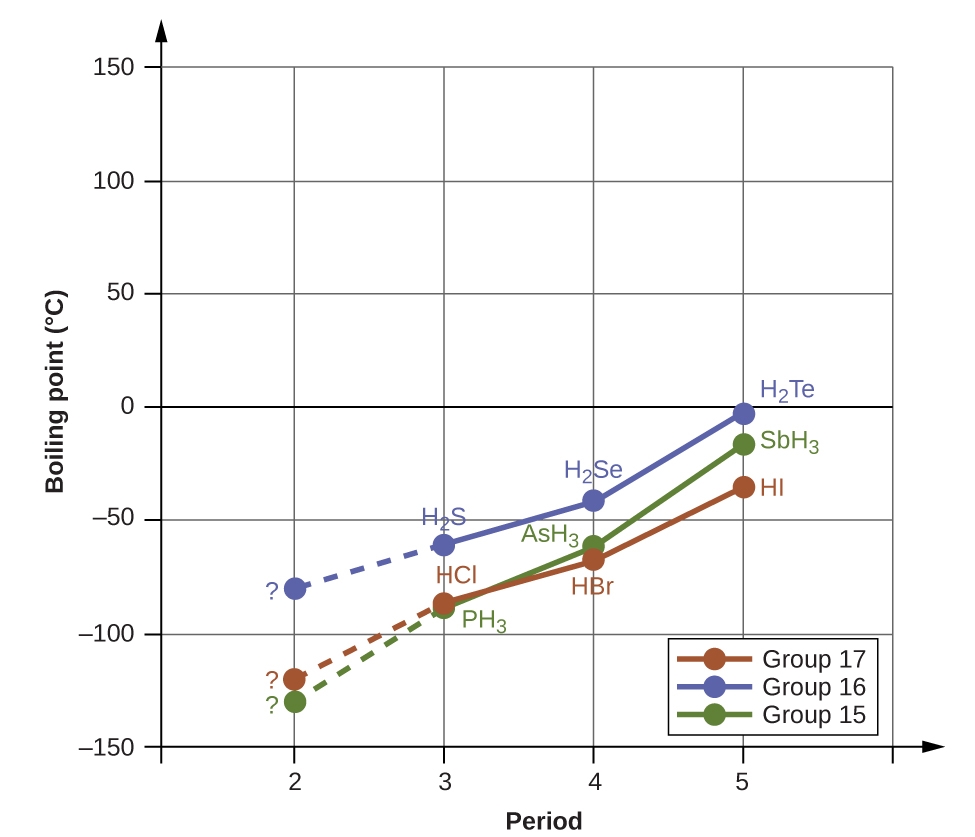
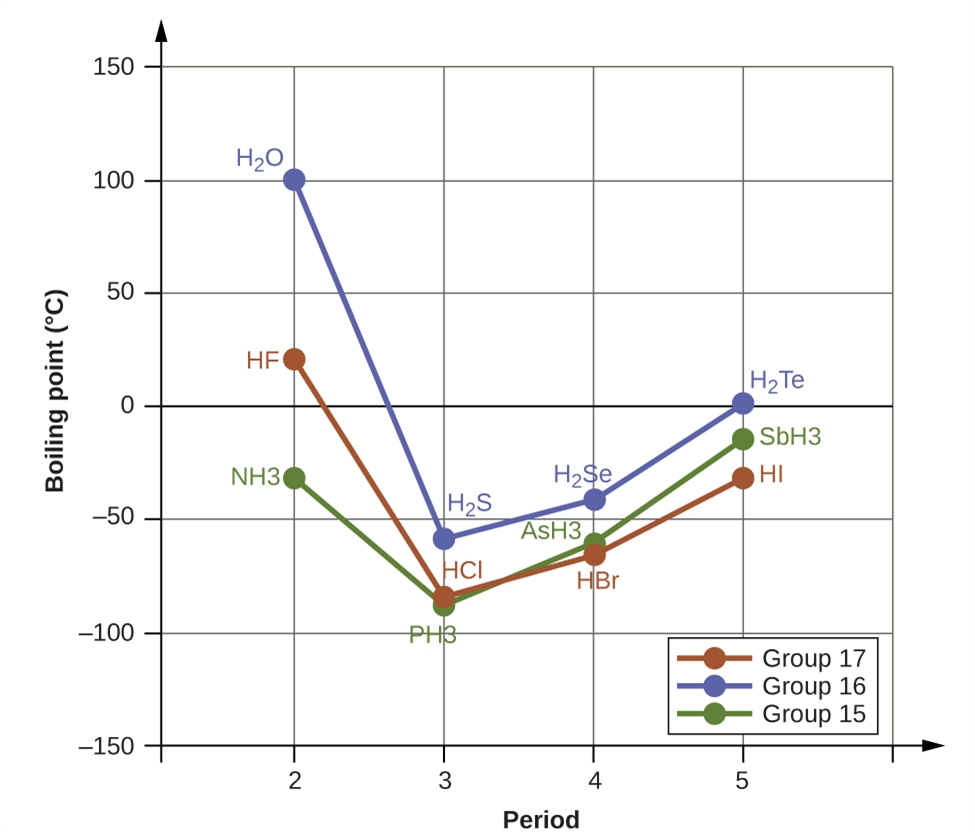
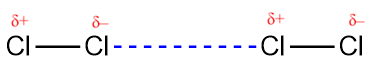

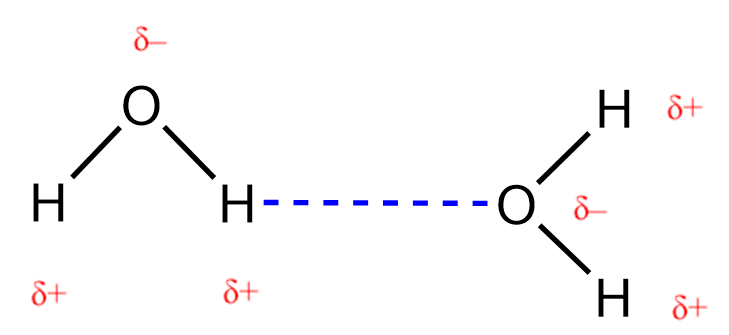
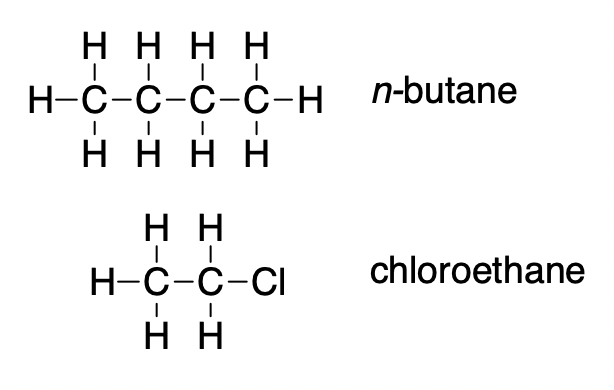
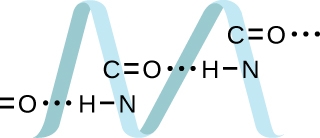
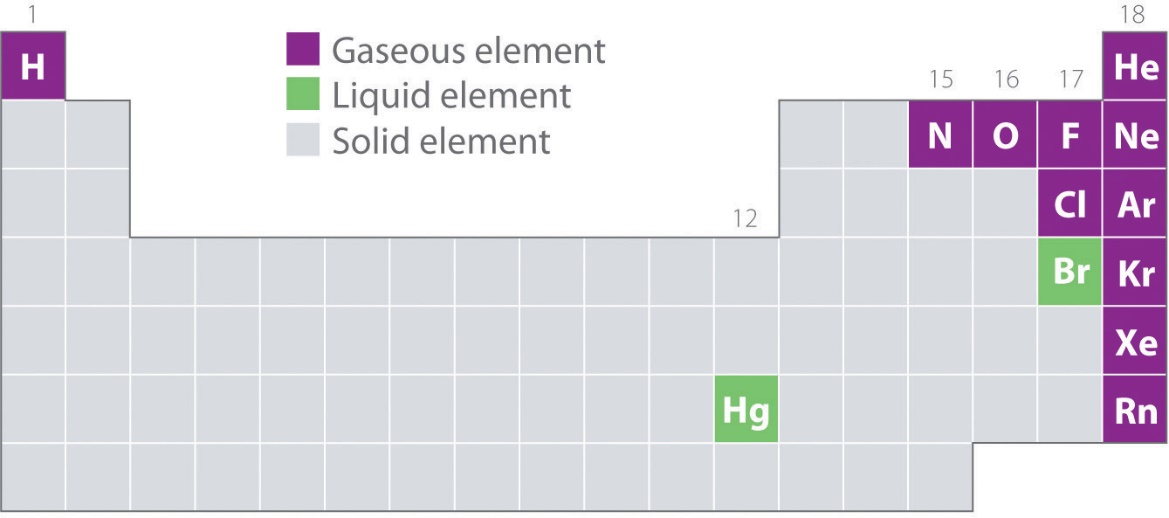
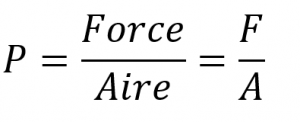
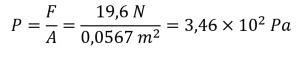
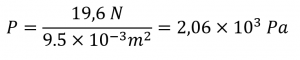

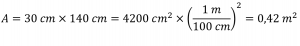
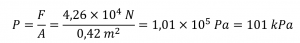
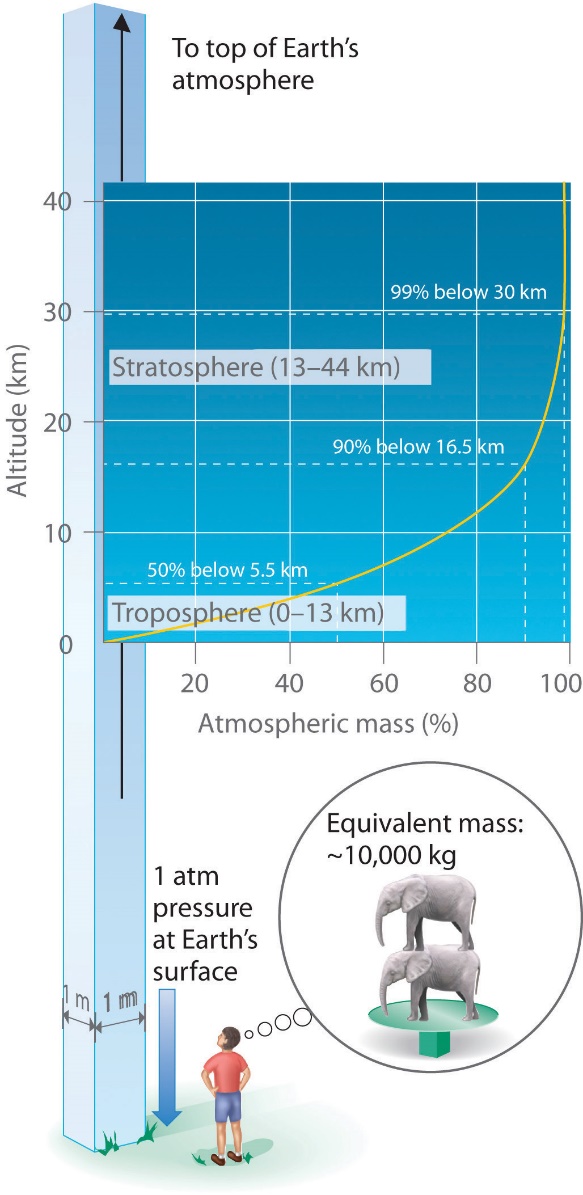
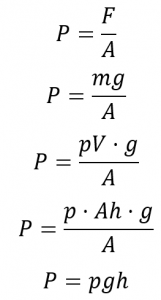
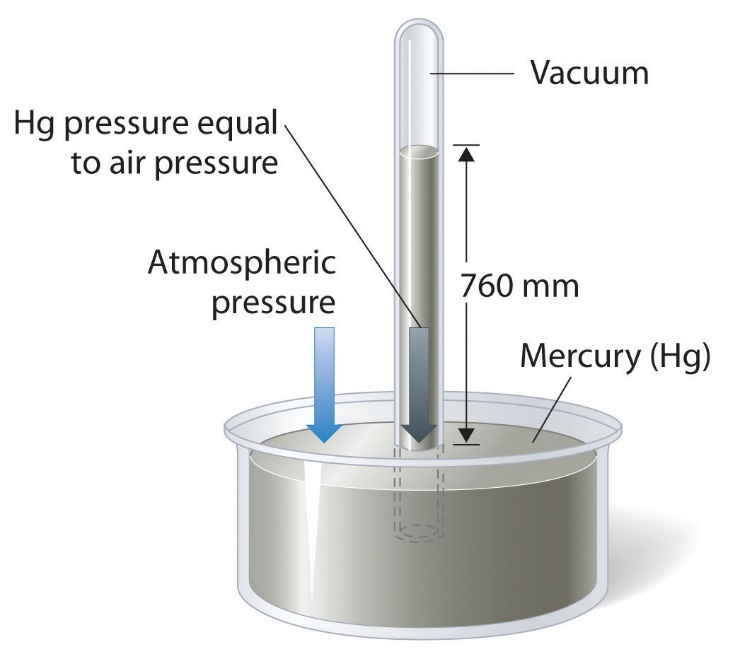
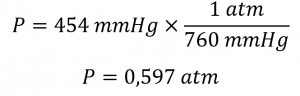
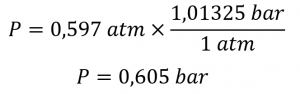
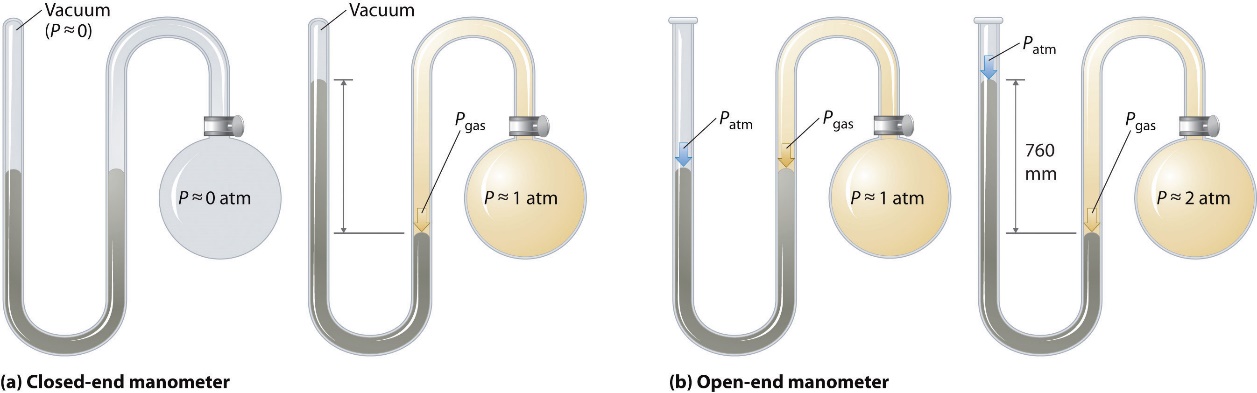
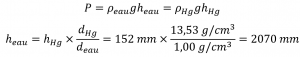
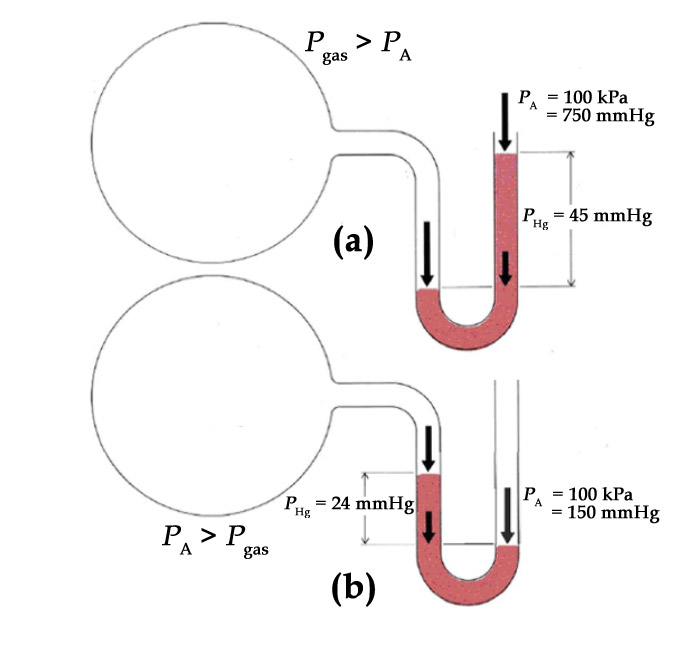
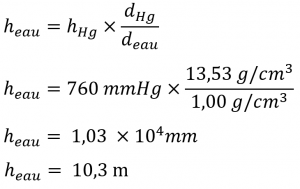
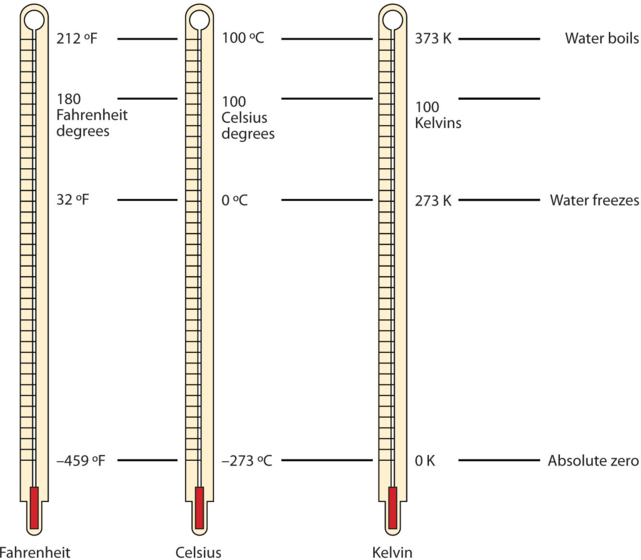
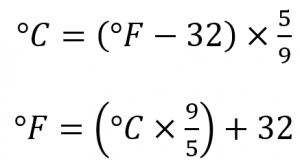
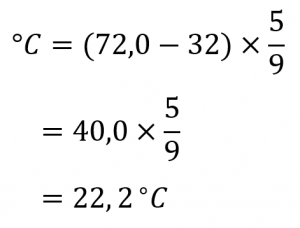
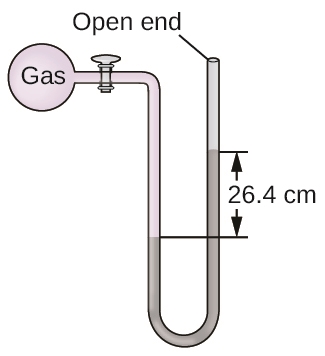
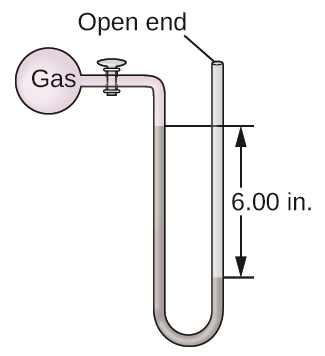

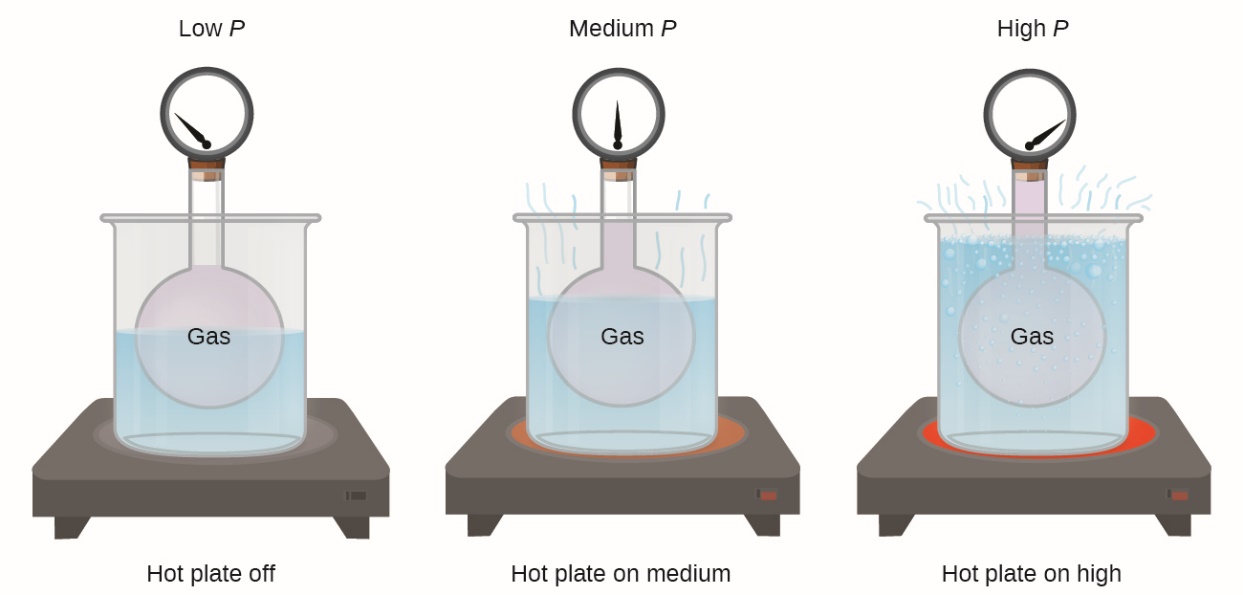
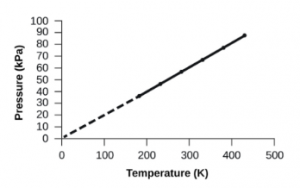
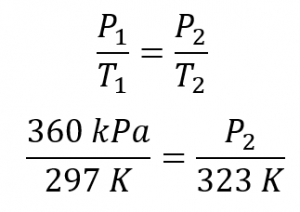
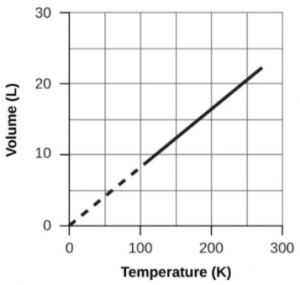
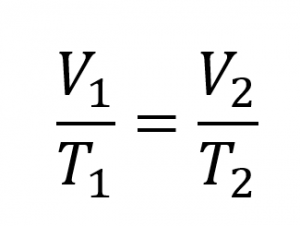
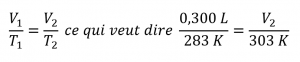
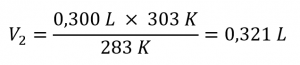
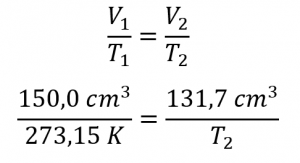
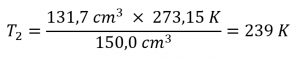
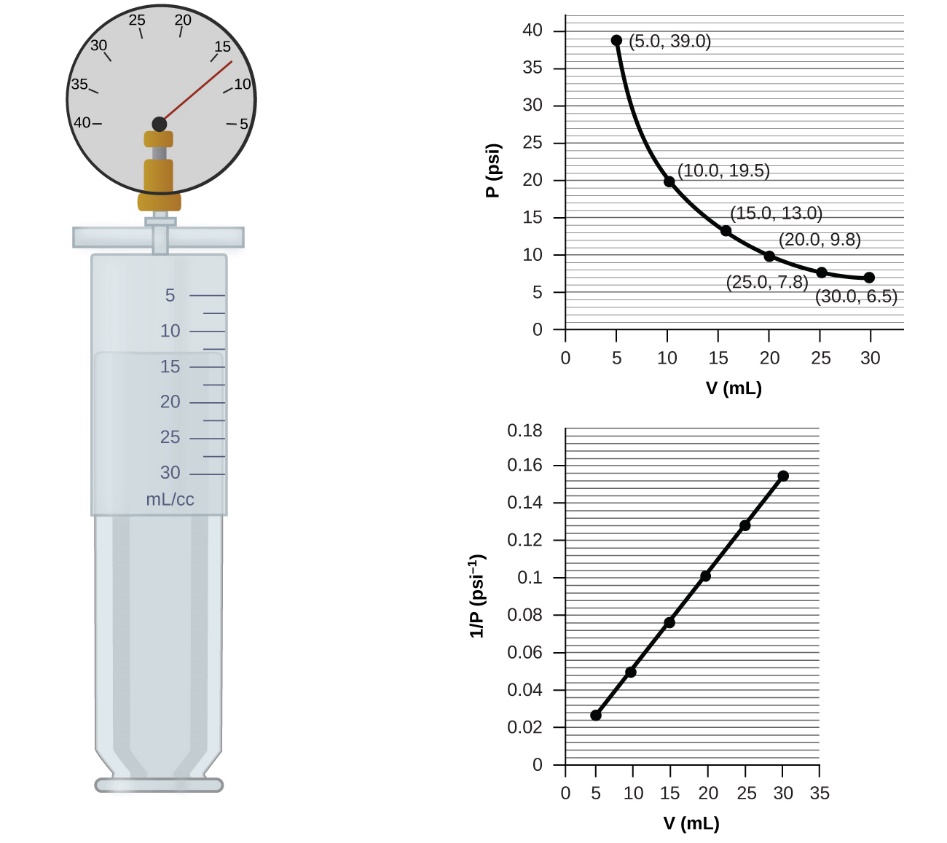
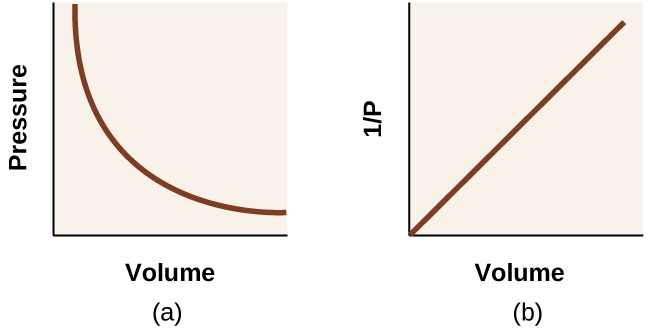
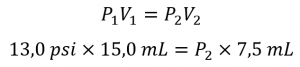
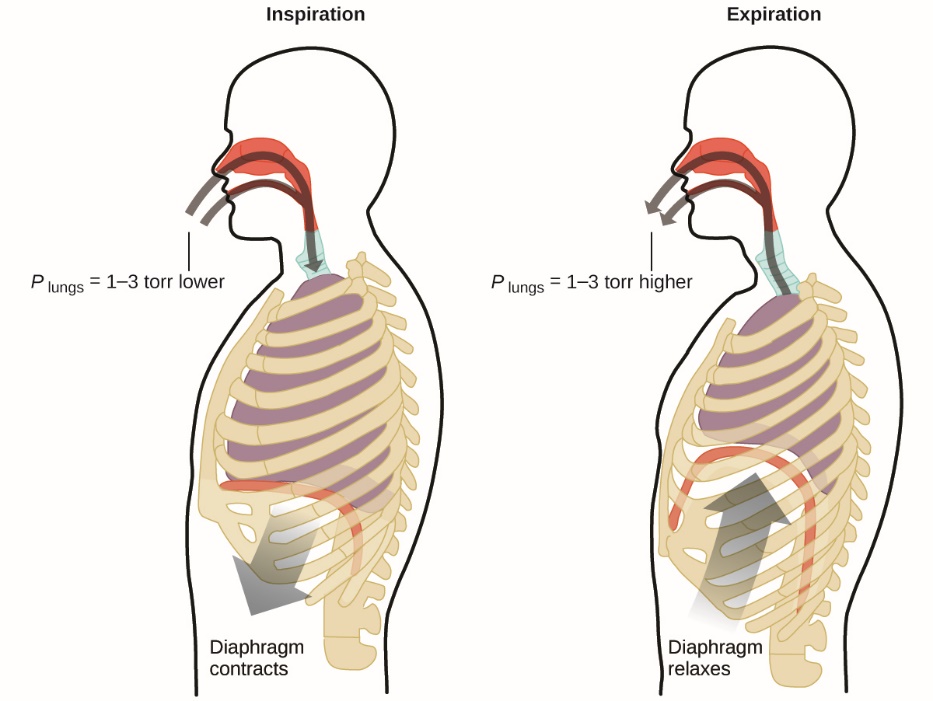

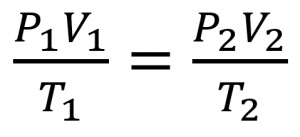

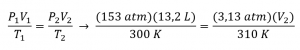
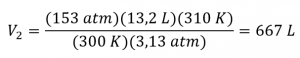

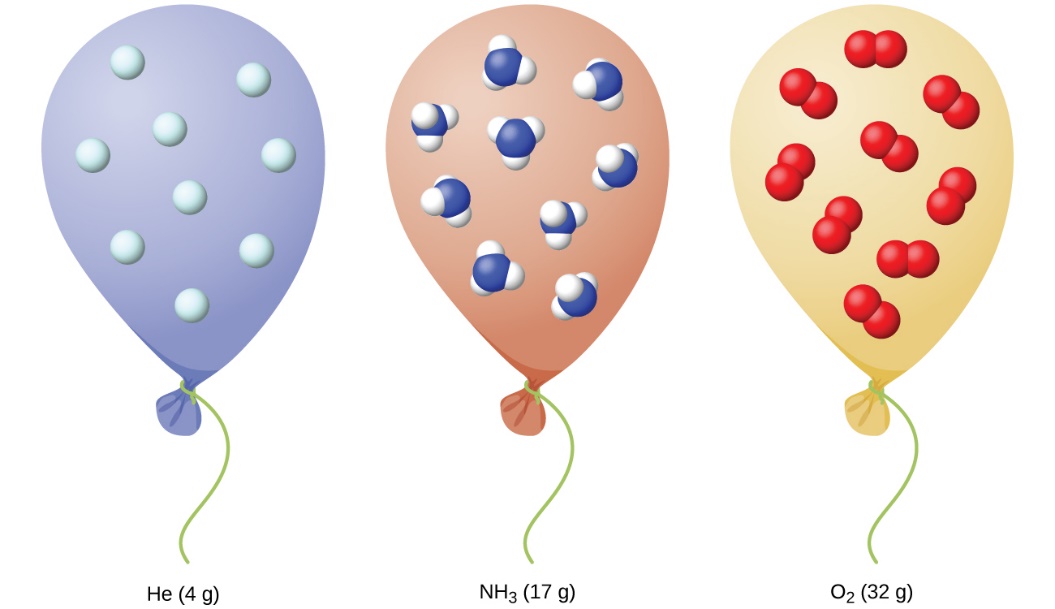

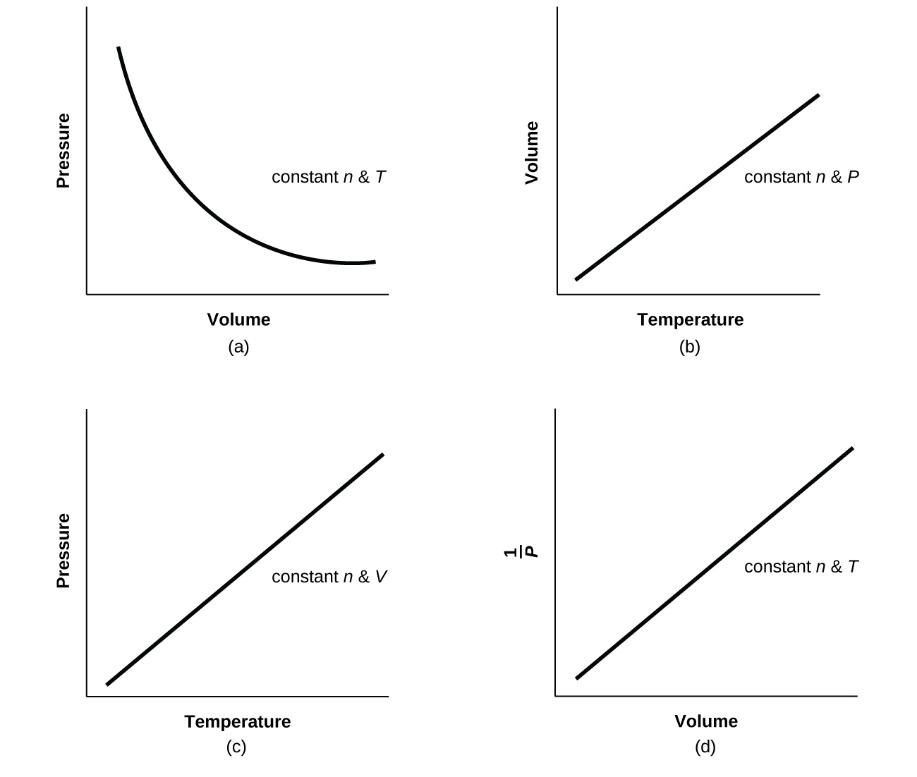
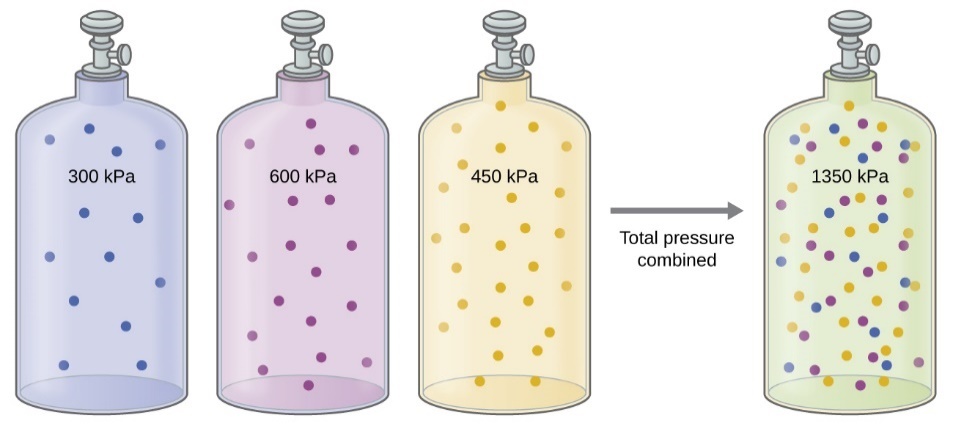
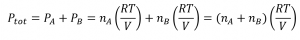
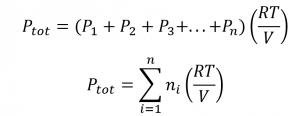
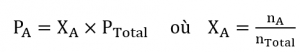

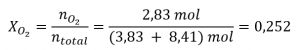
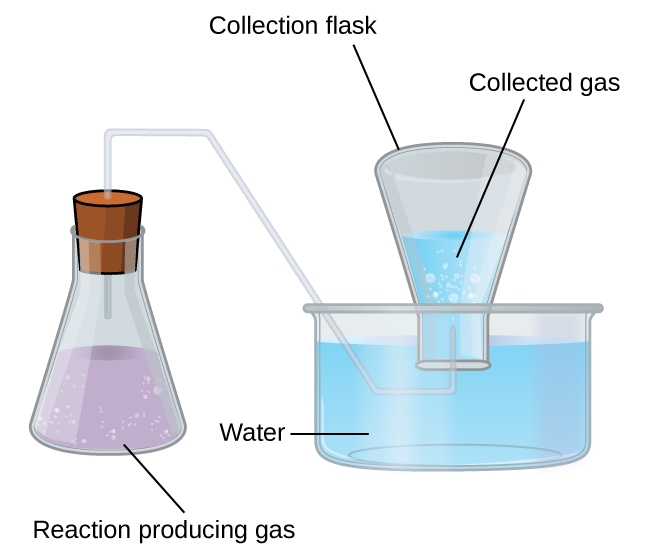
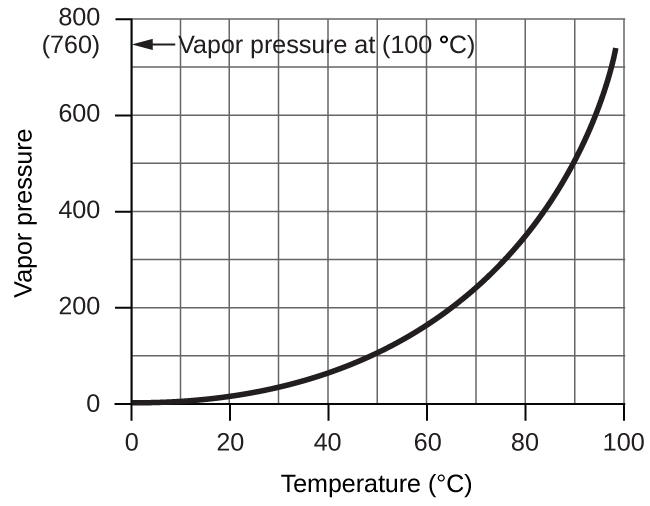

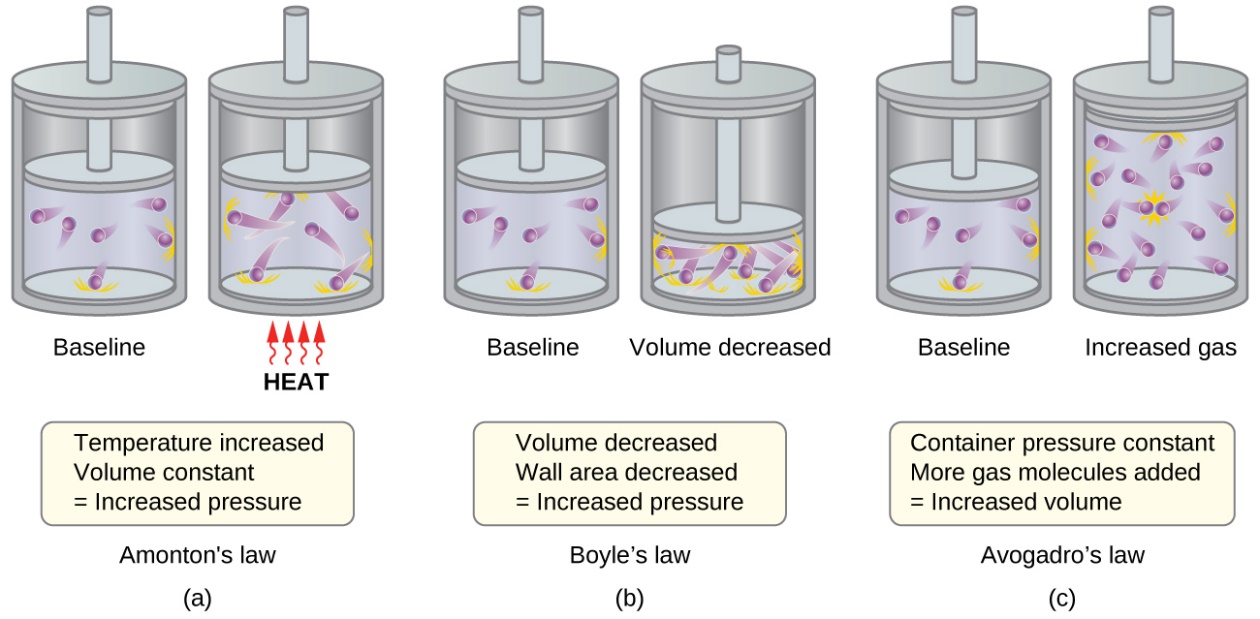
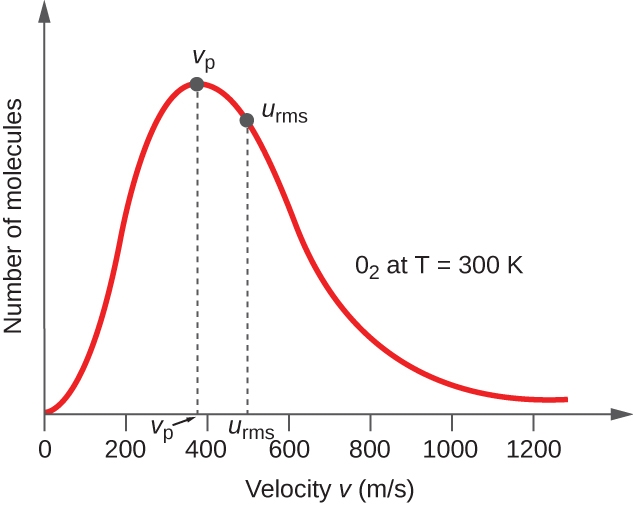
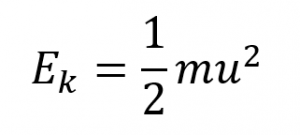
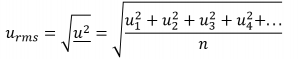
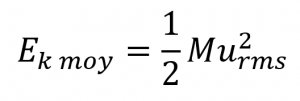
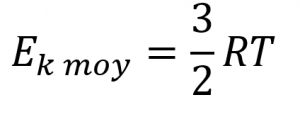
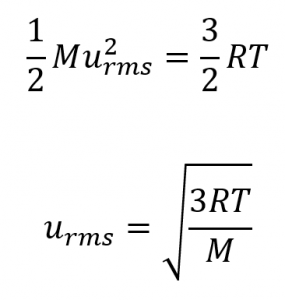
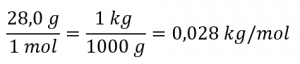
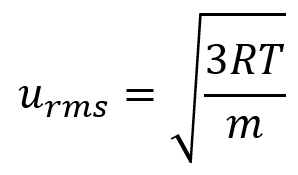
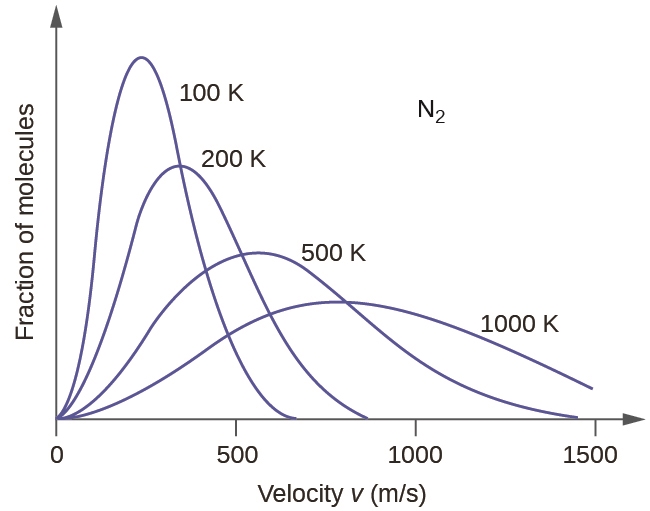
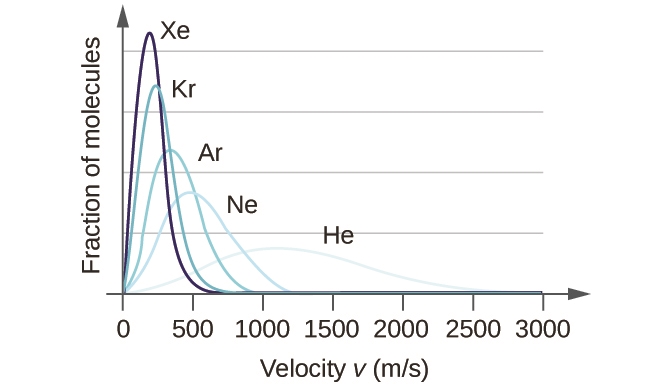
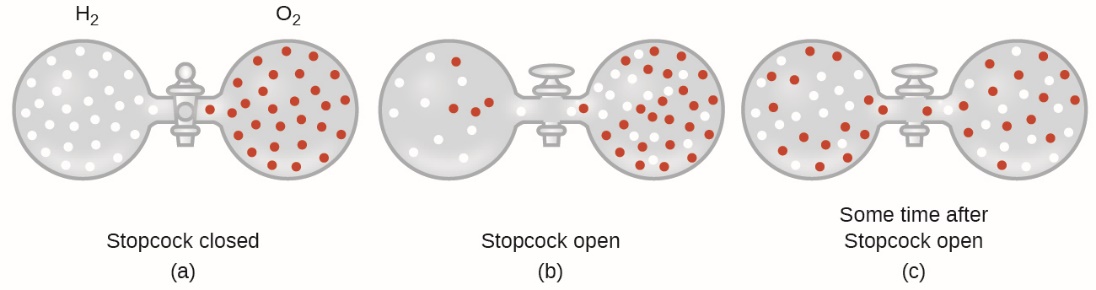
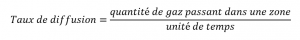
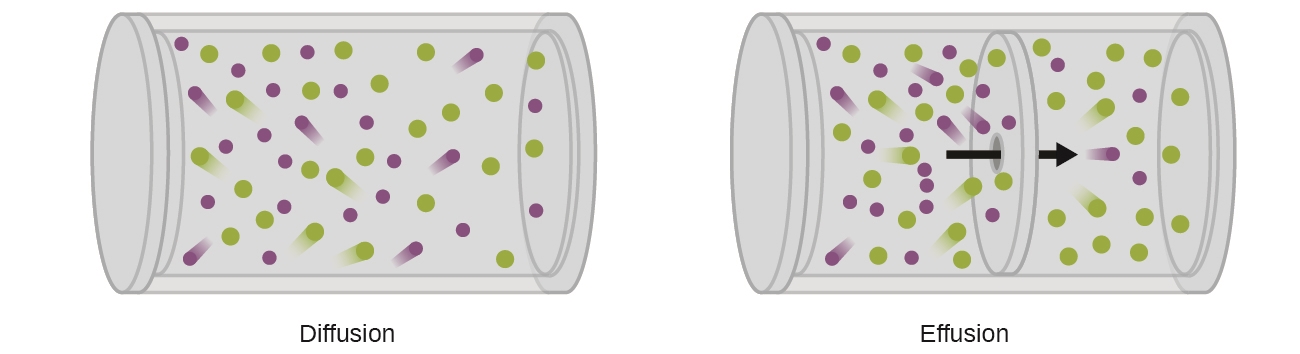
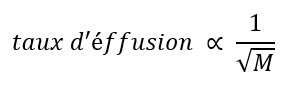
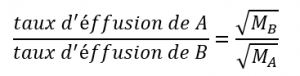
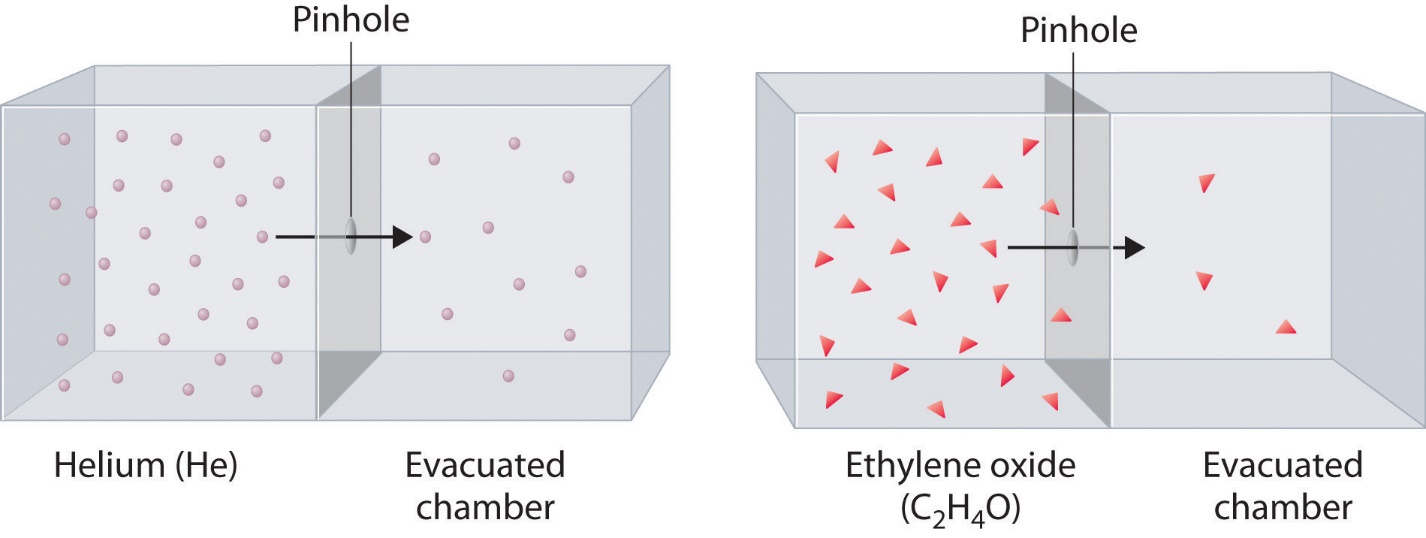
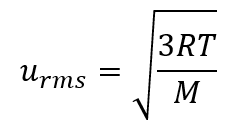
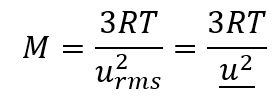
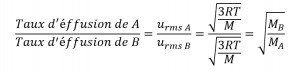
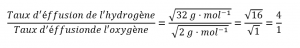
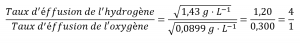
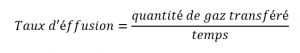


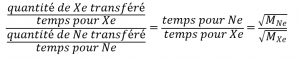
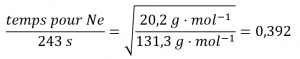
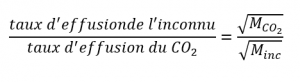
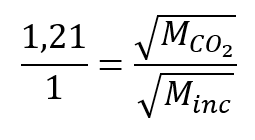
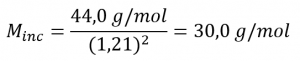
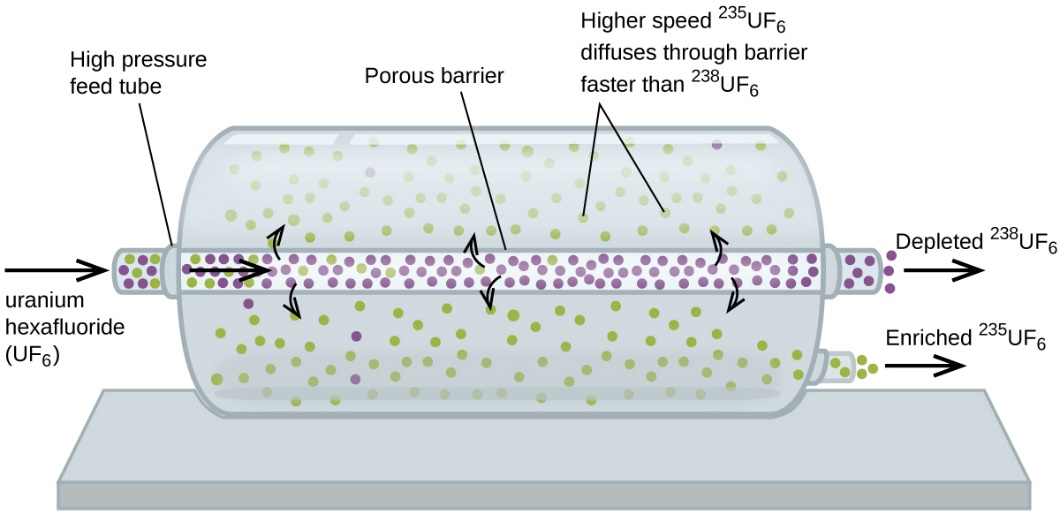
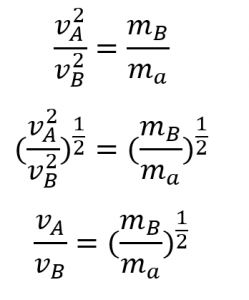
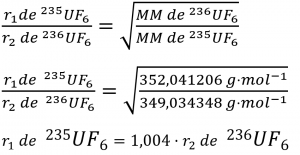
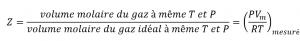
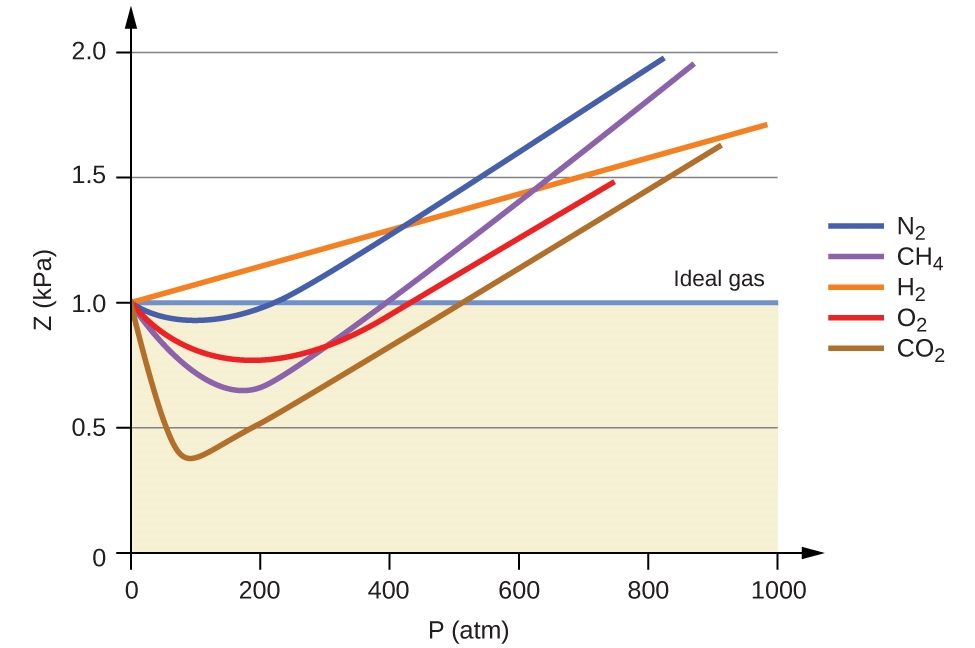
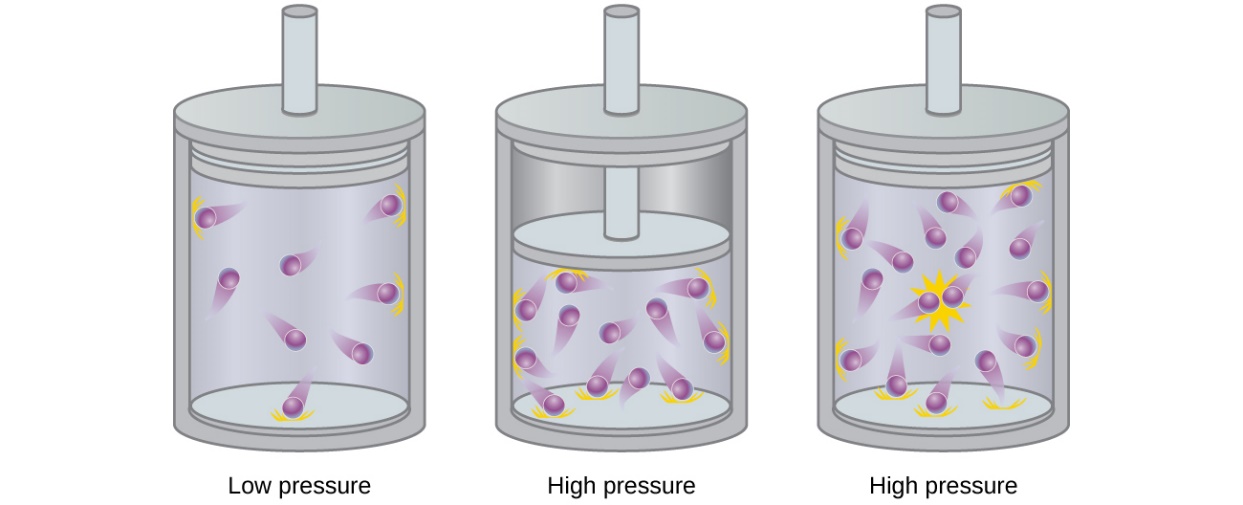
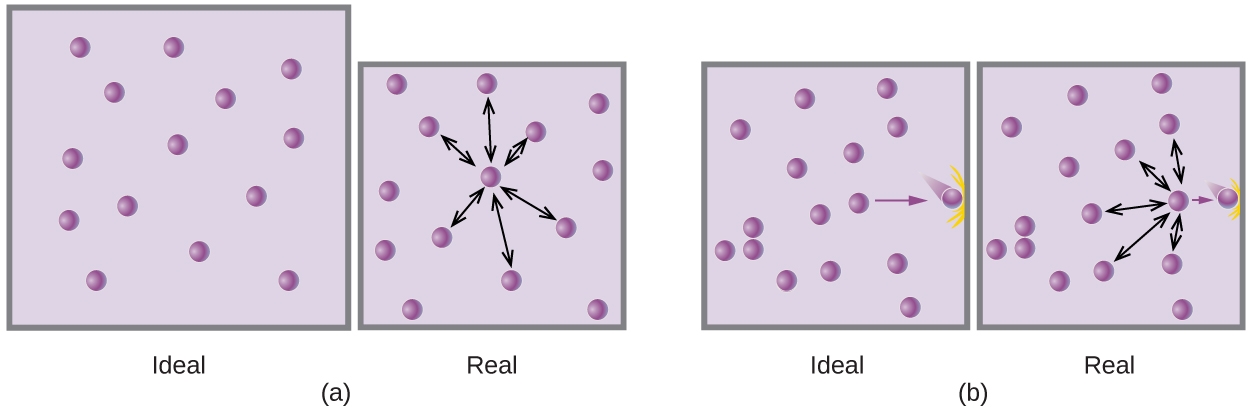
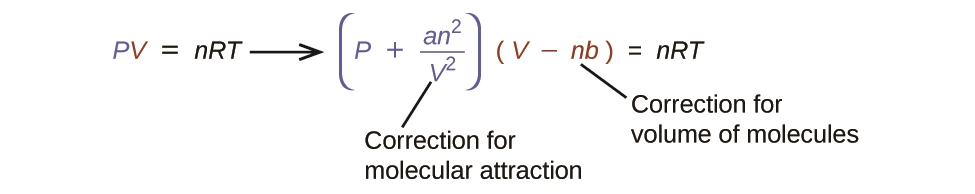
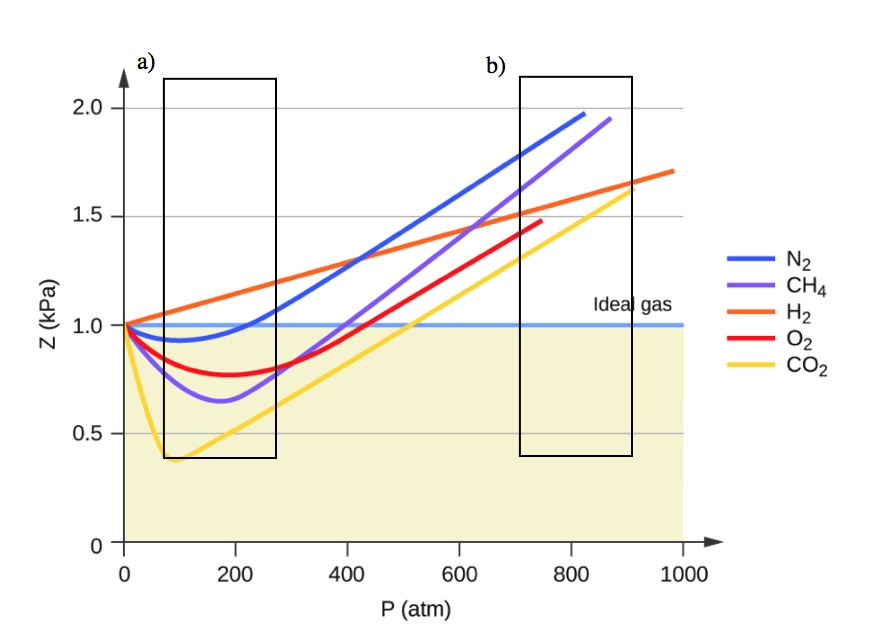
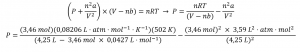
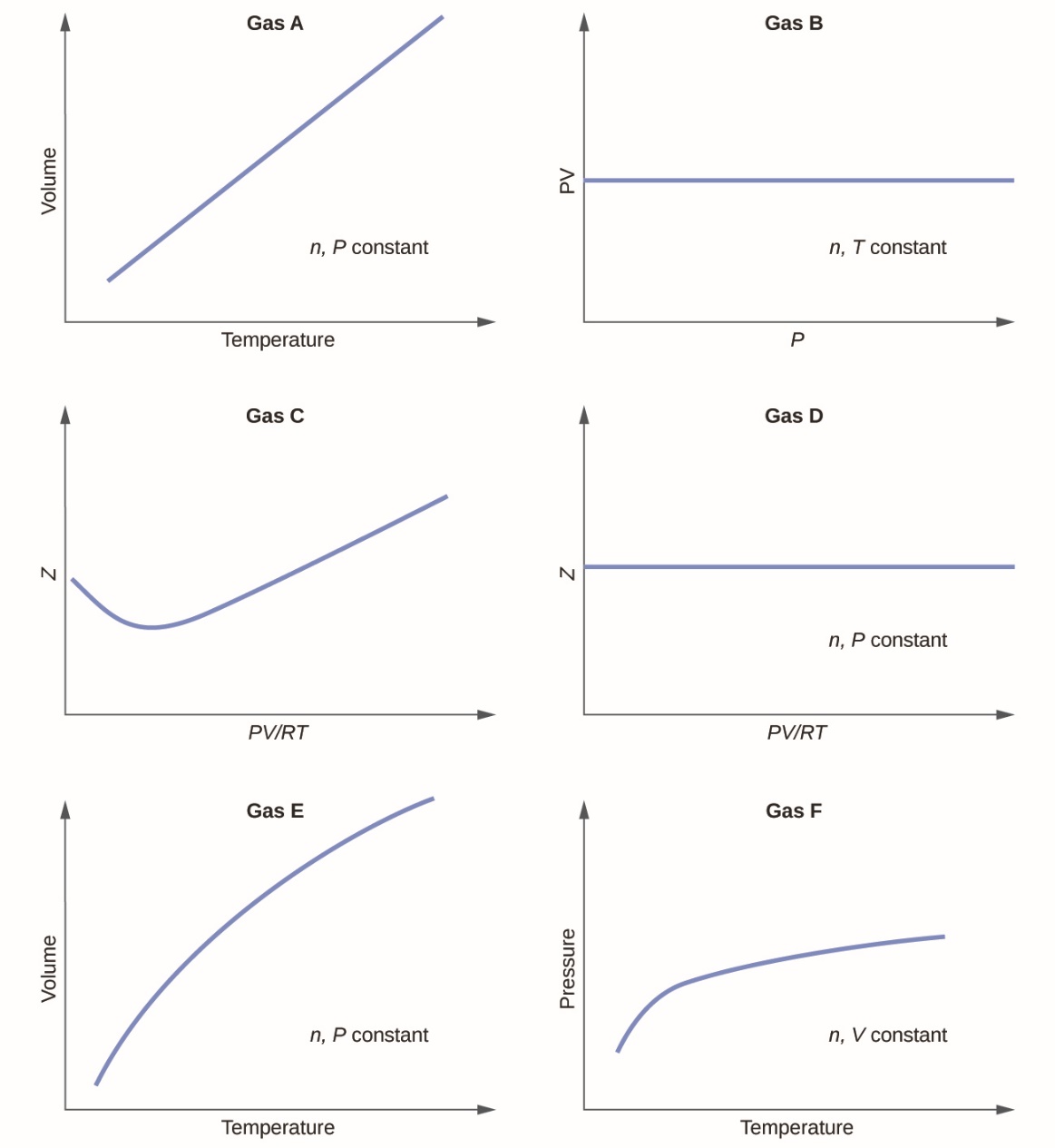
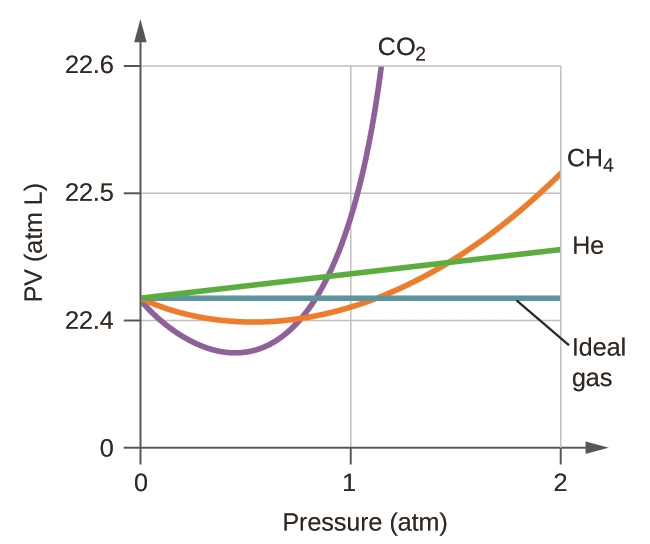


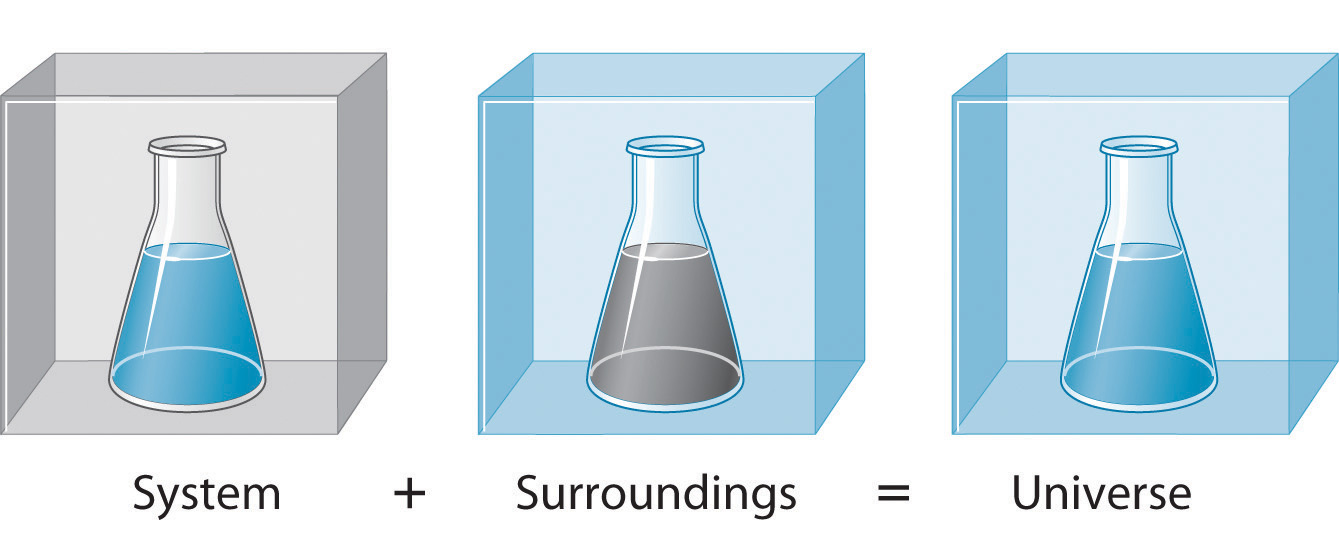
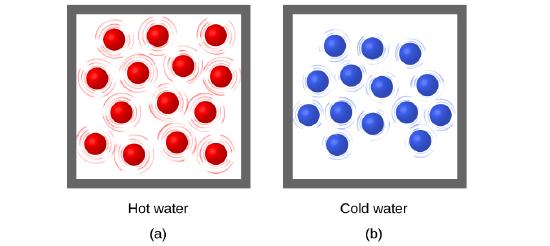
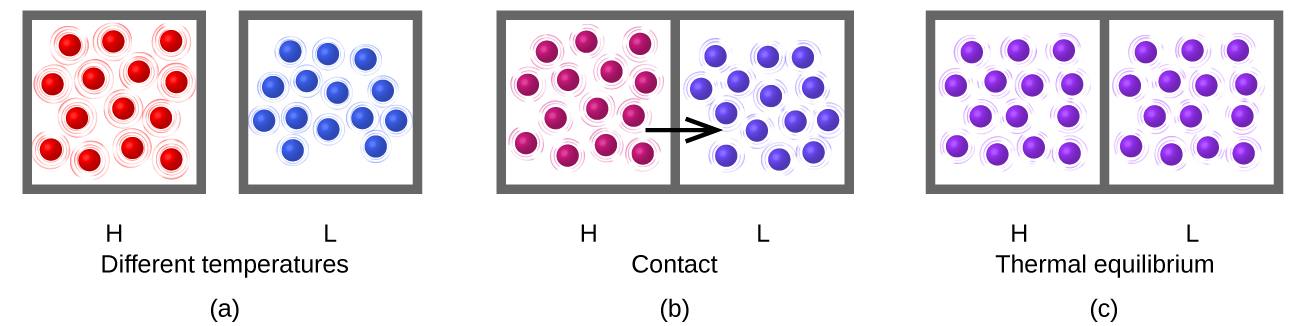



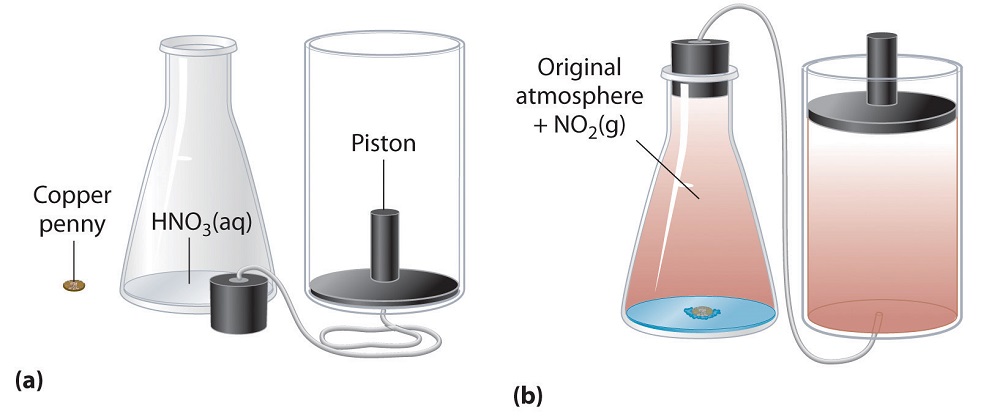
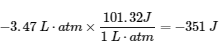
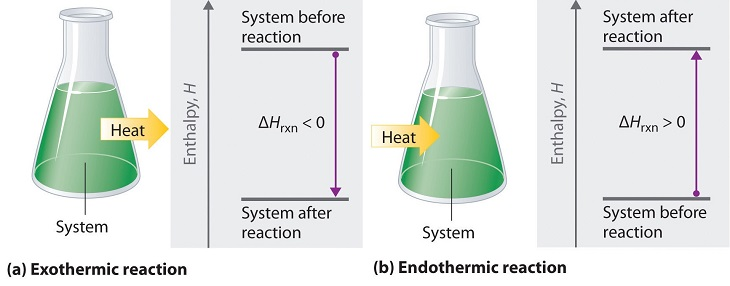
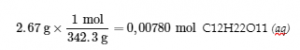
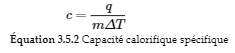
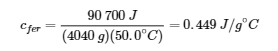

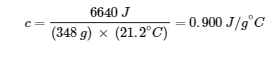
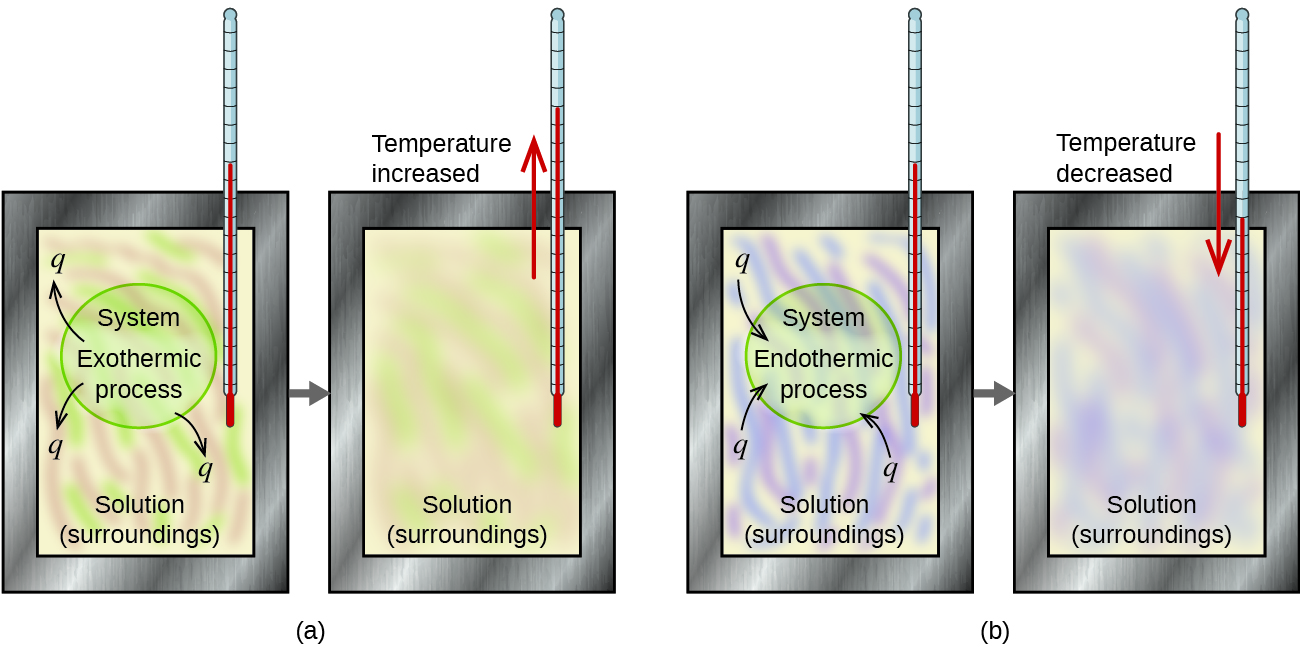
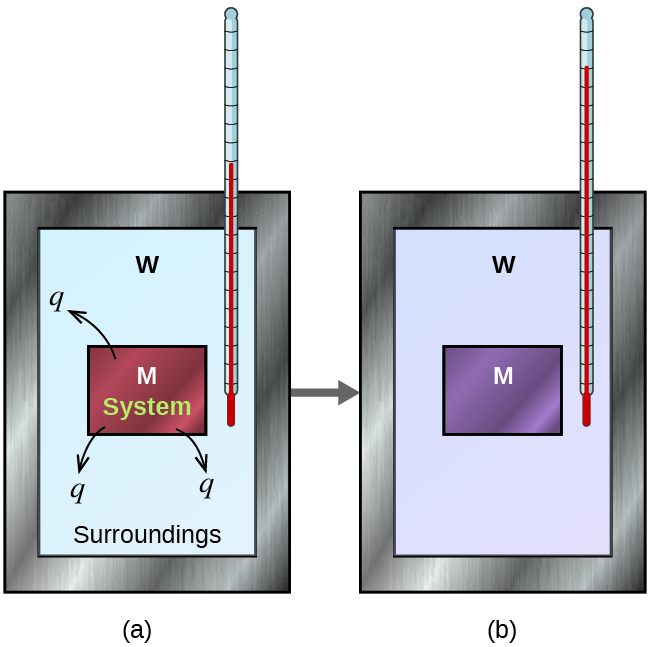
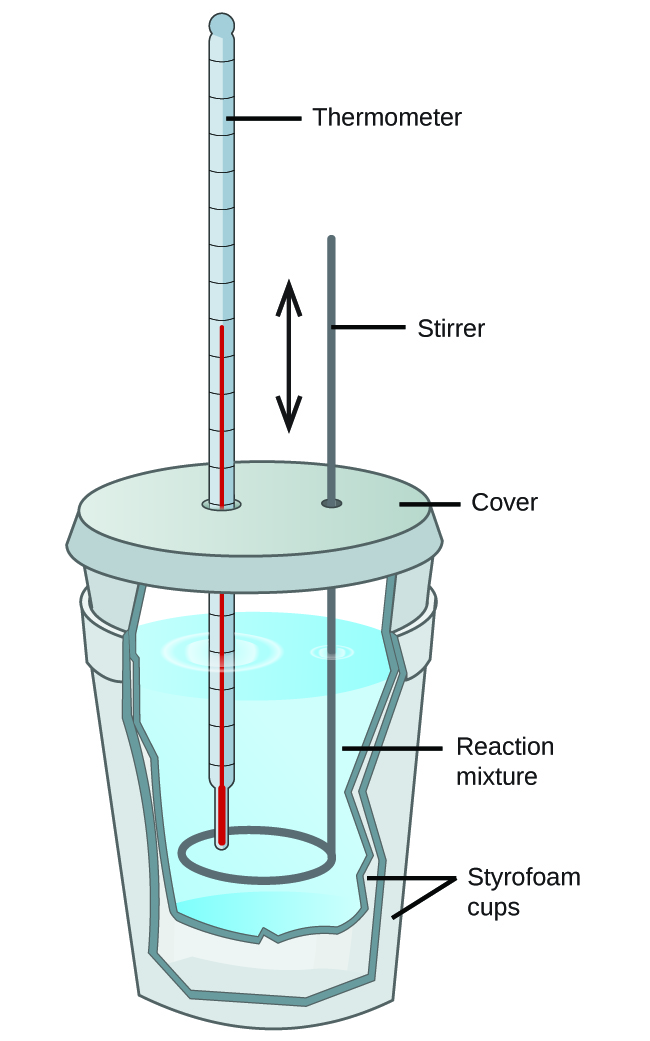
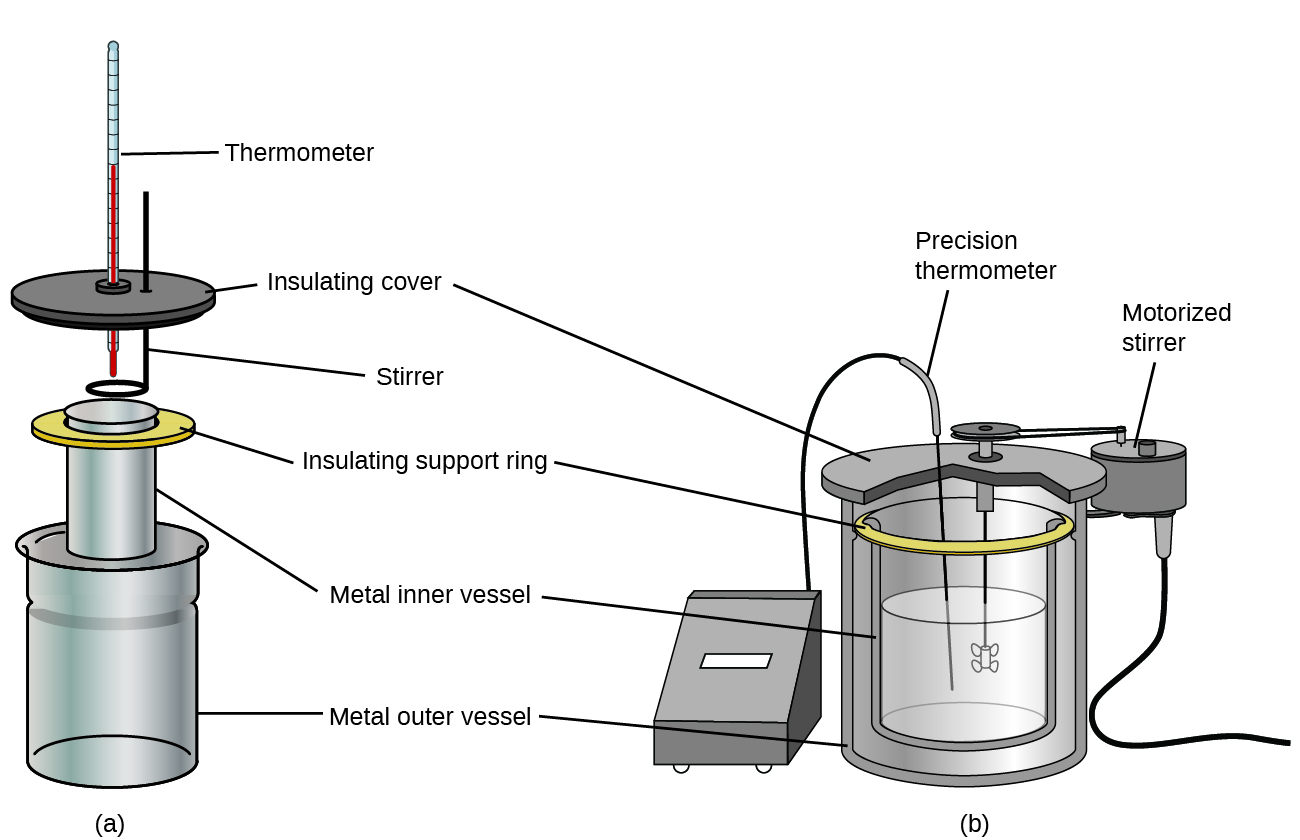
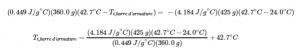


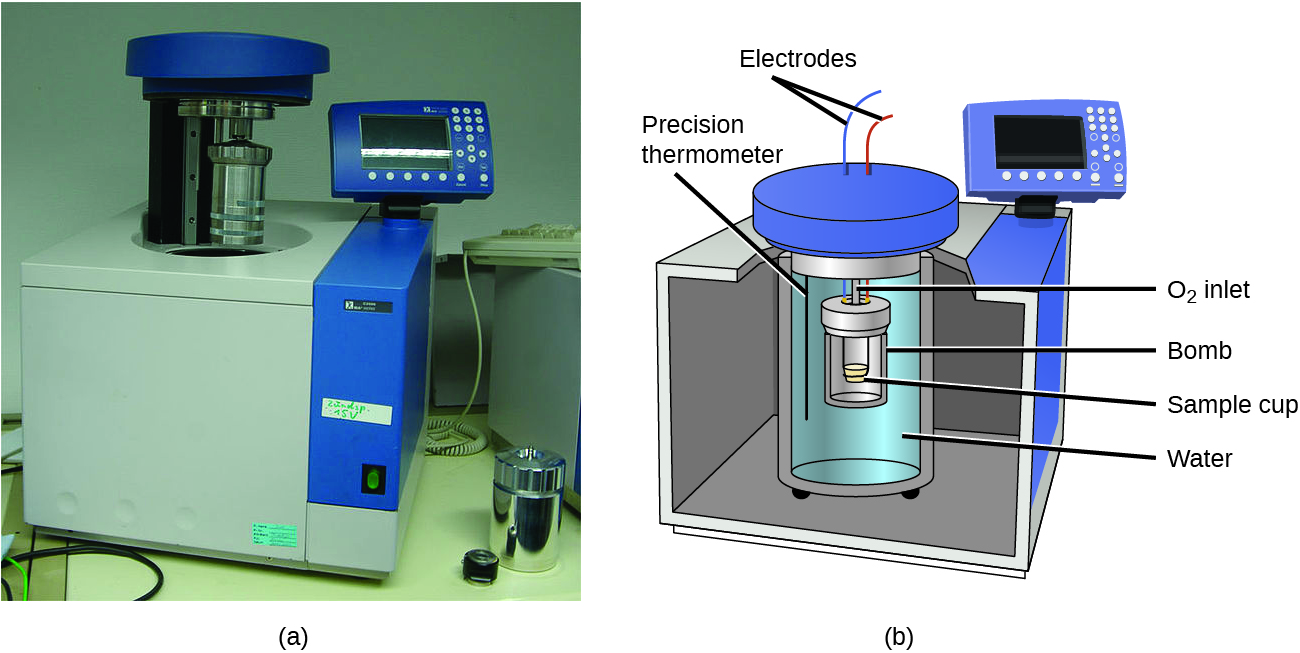

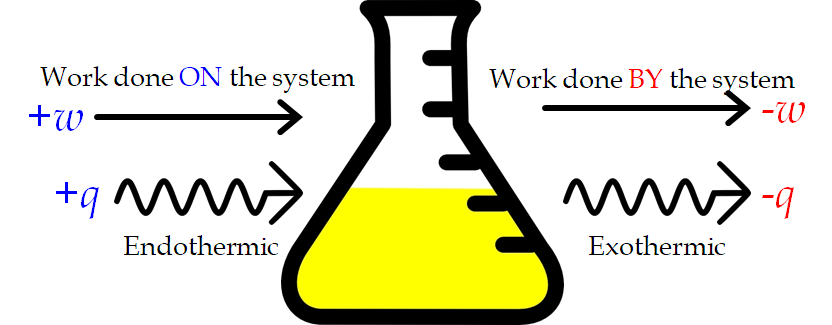
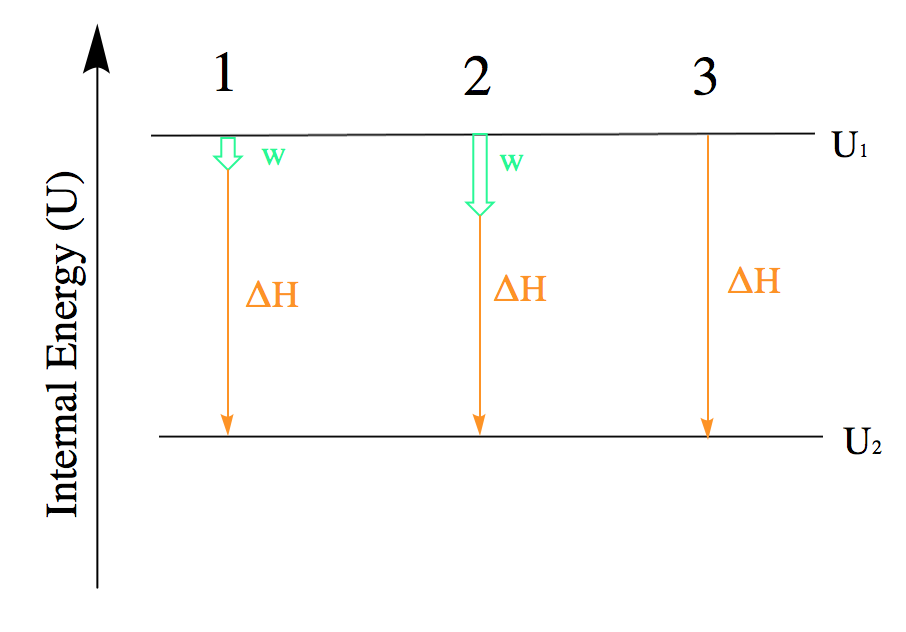
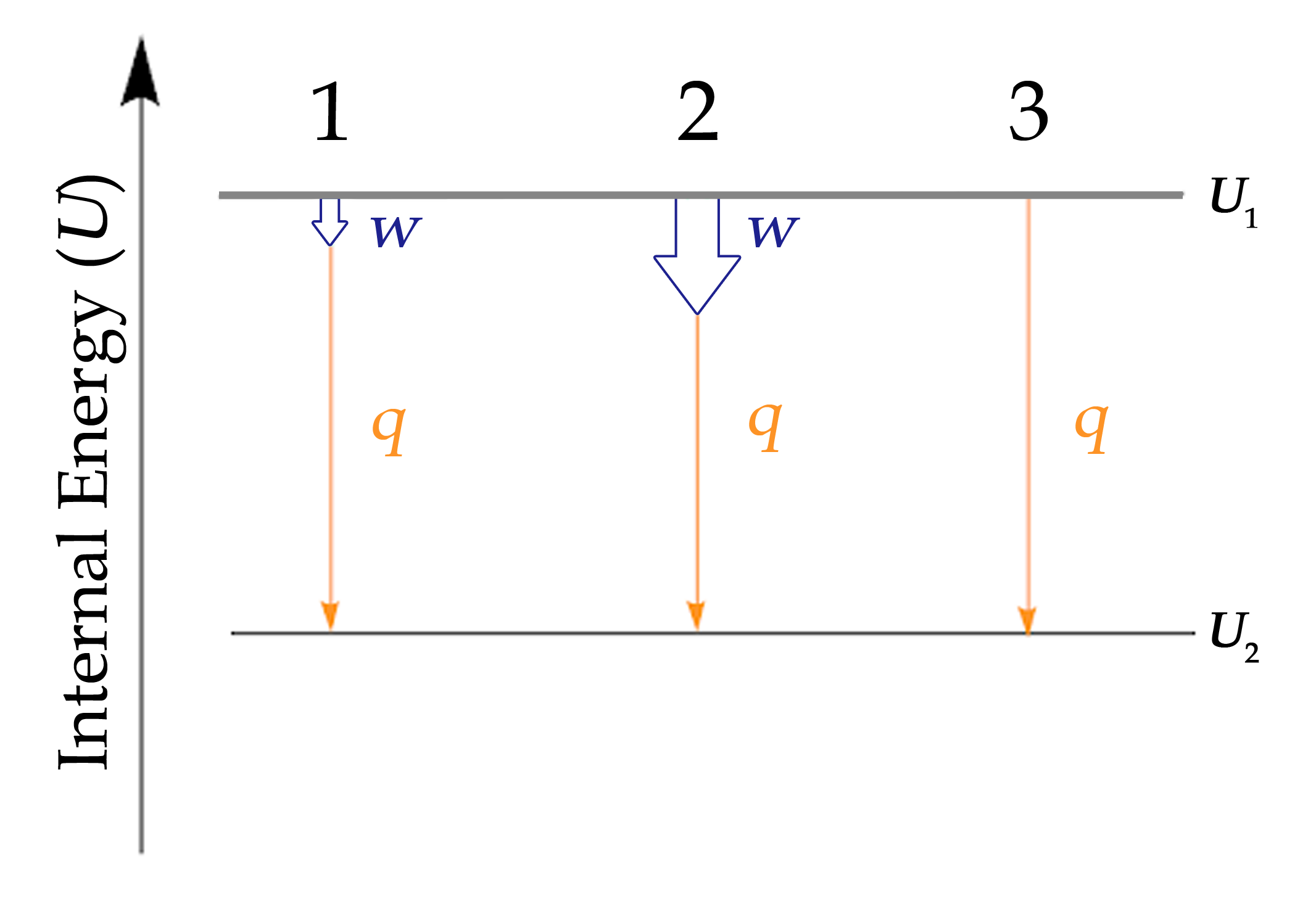
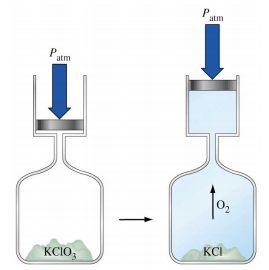
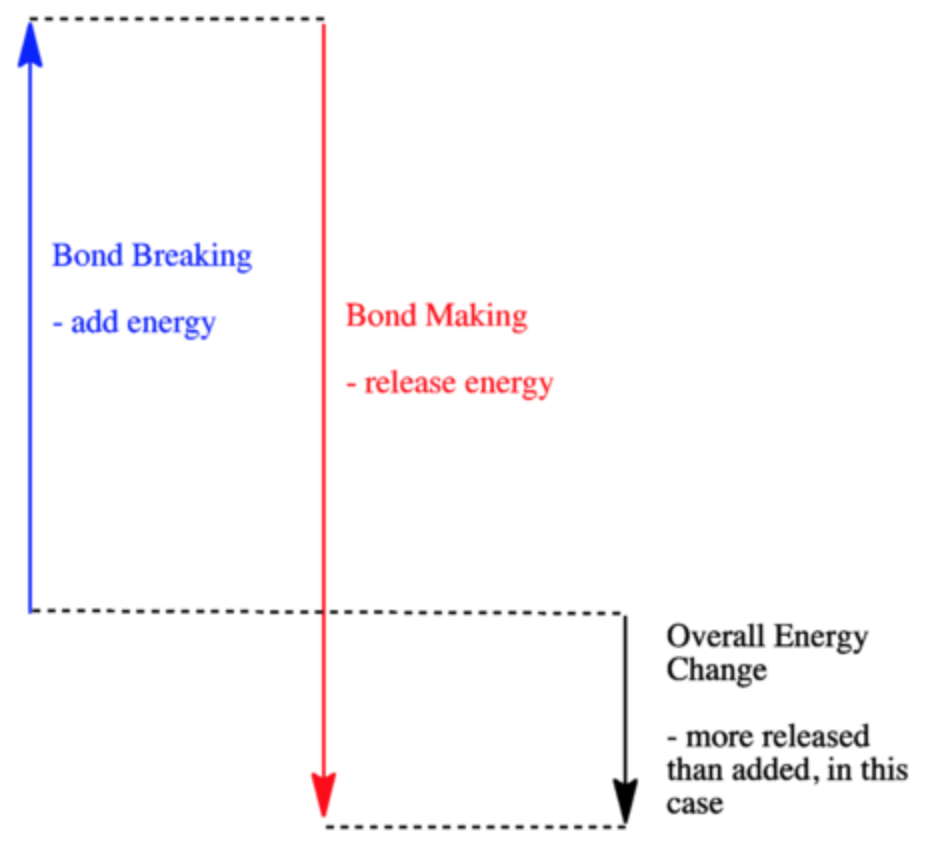

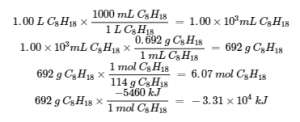


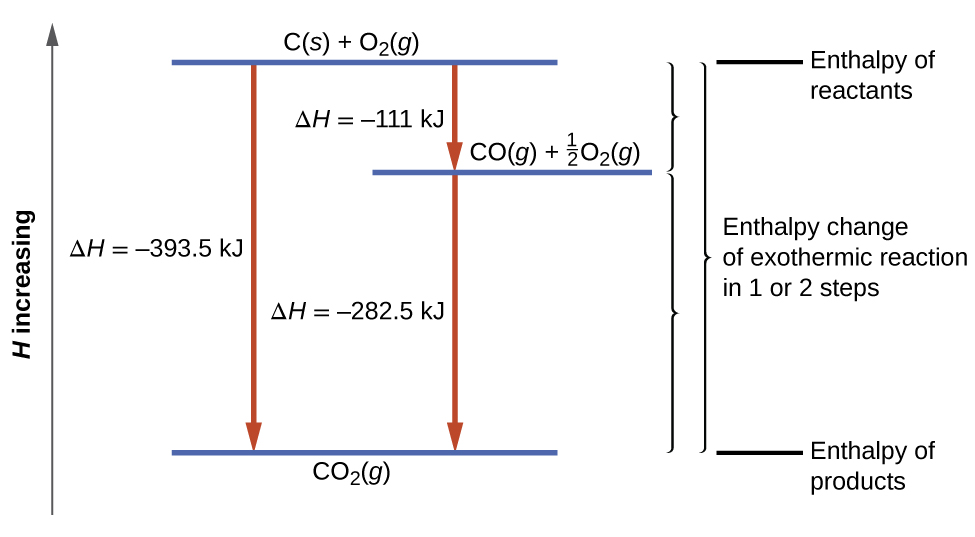
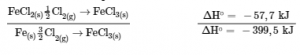

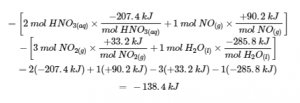
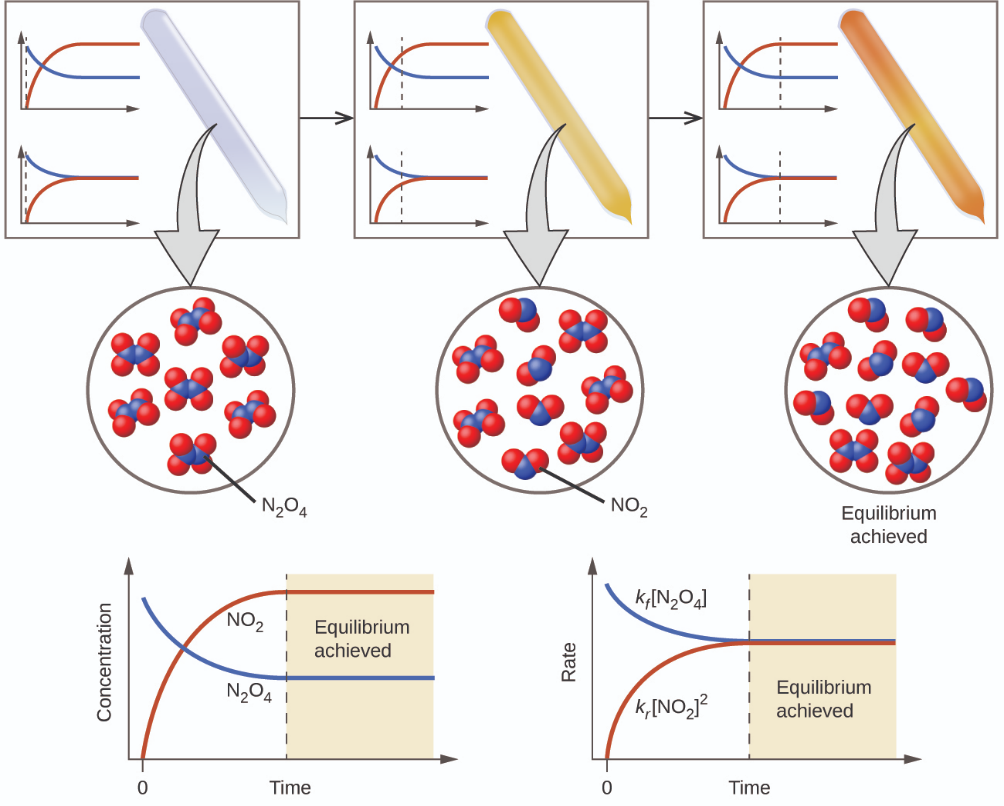



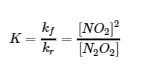
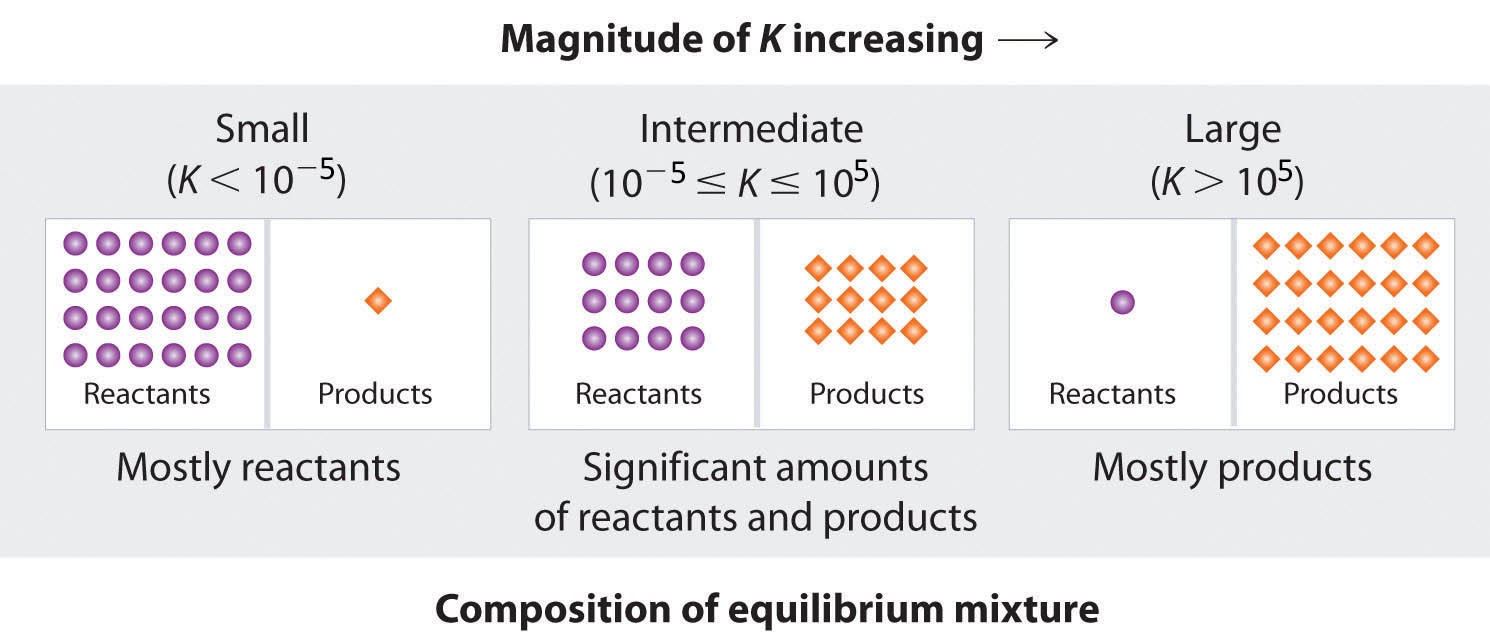
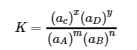
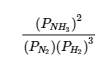
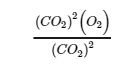
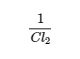
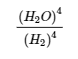
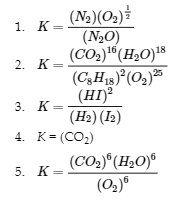
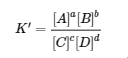
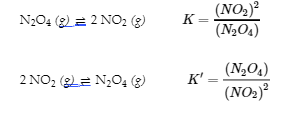
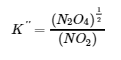
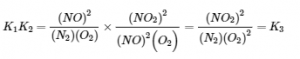
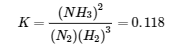
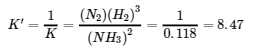
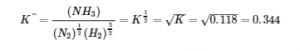
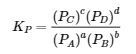
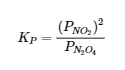
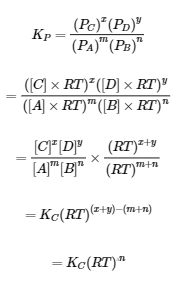

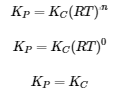
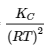
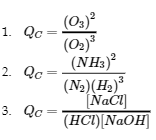
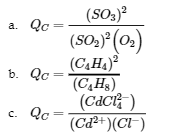
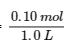
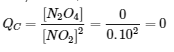
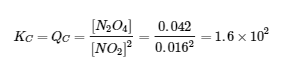
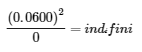
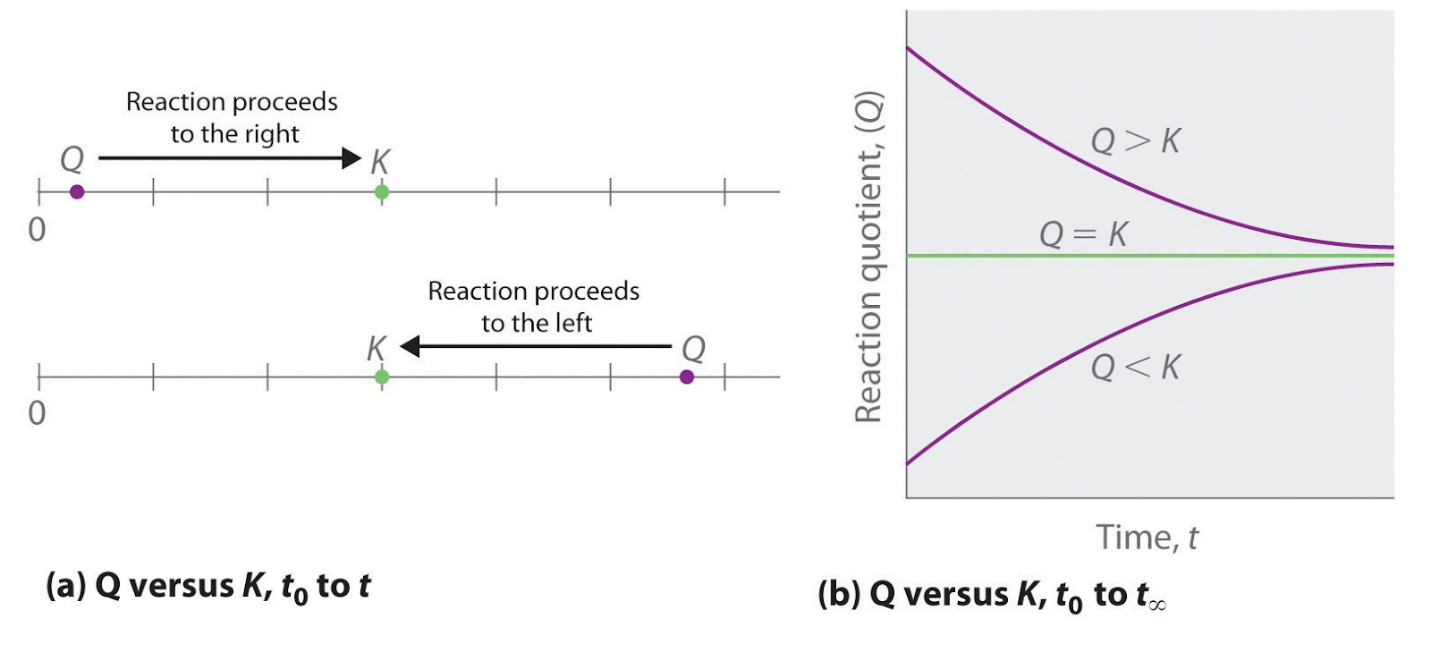
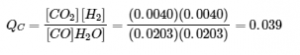
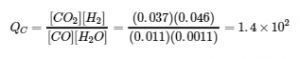
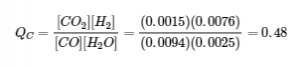
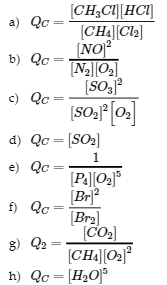
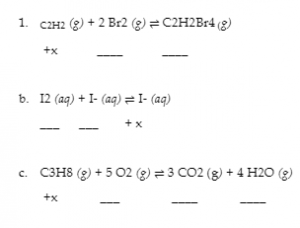
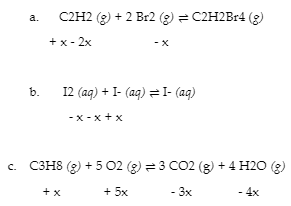
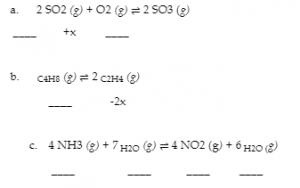
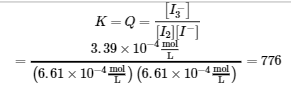
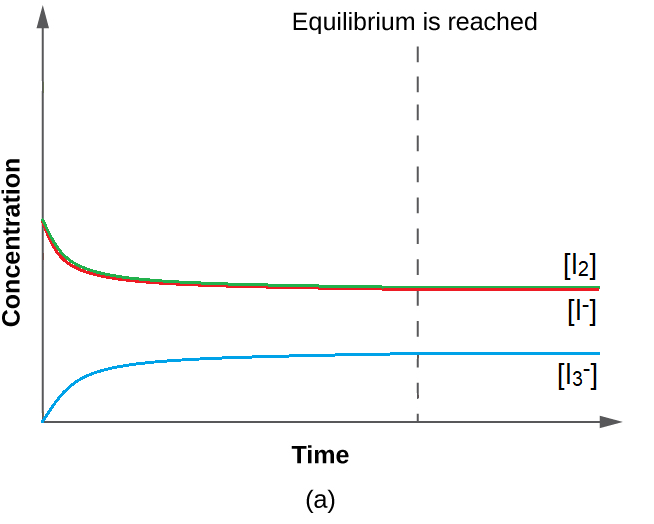
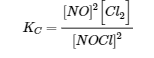


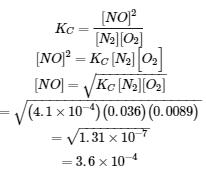
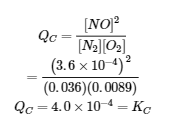
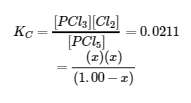
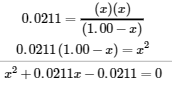

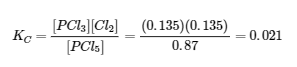
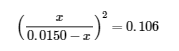
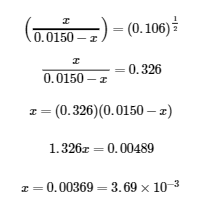
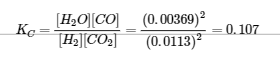
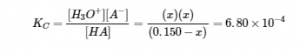
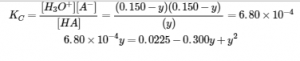
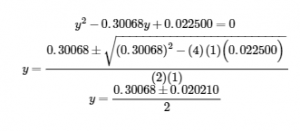
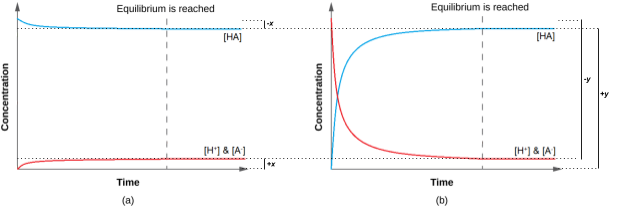
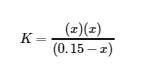
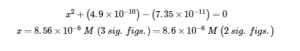
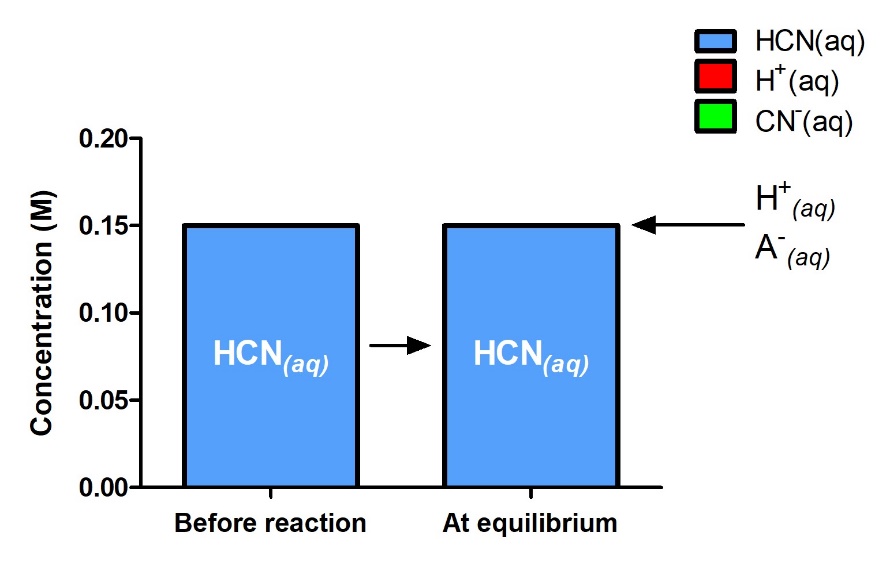
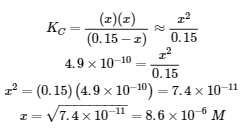
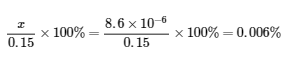
 C
C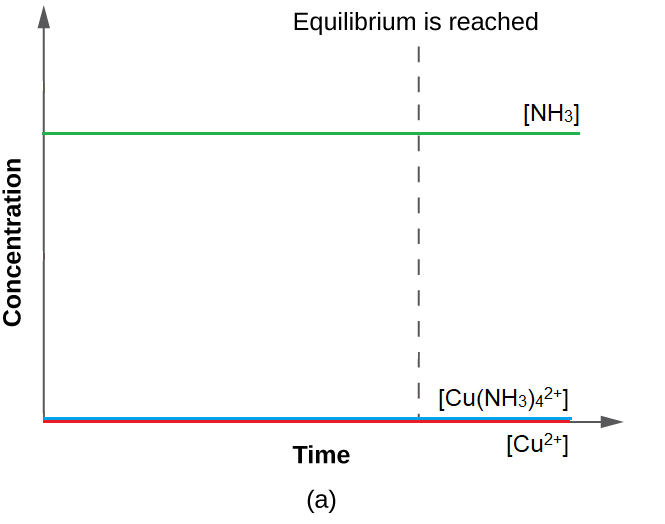
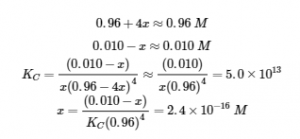
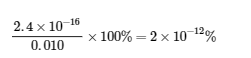

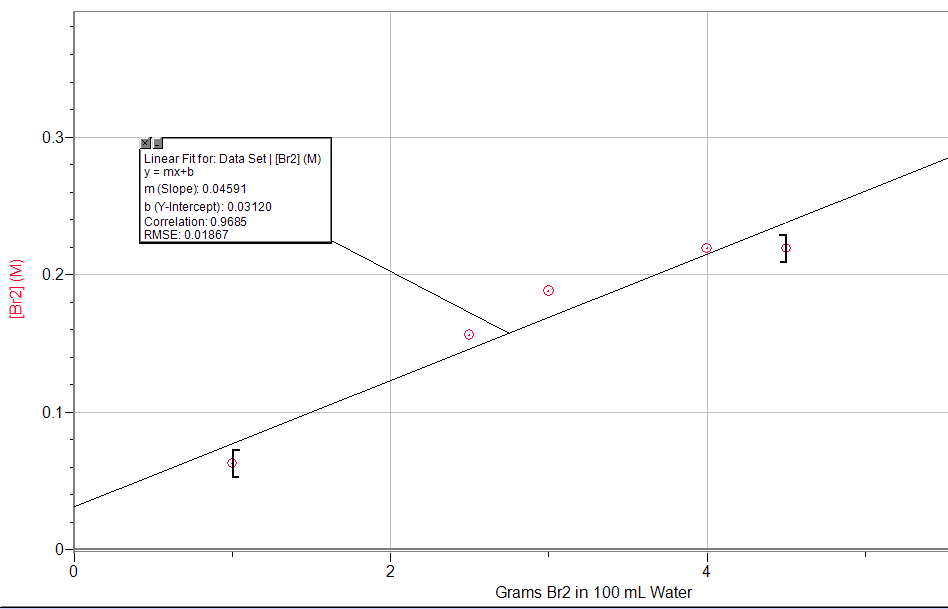


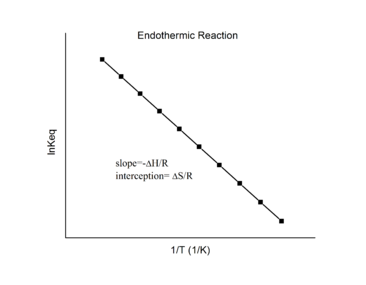
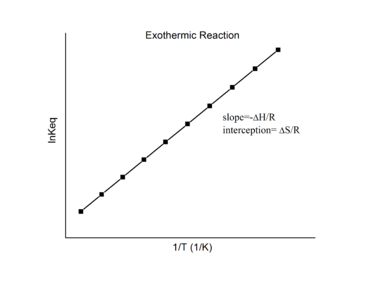


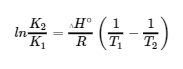
 L
L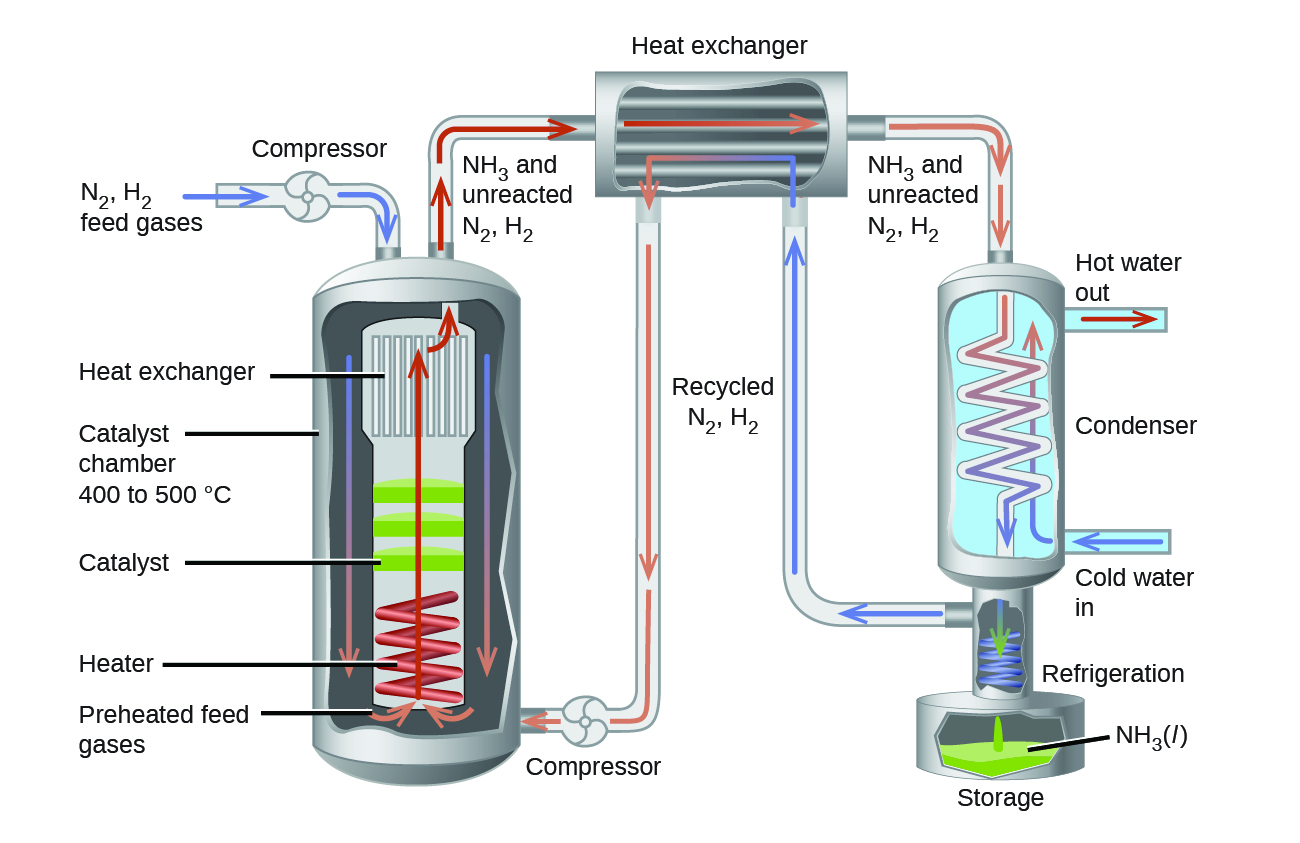

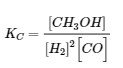
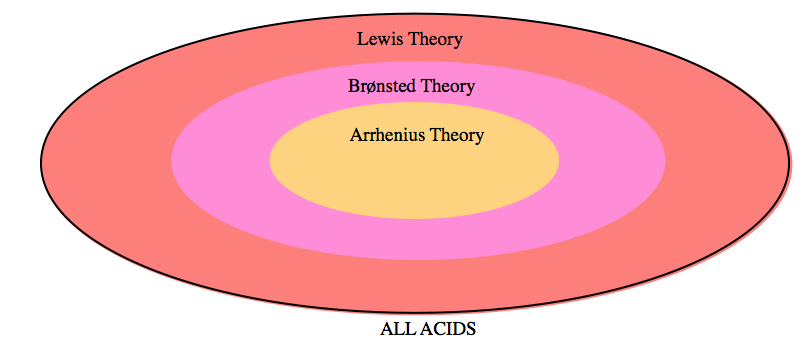
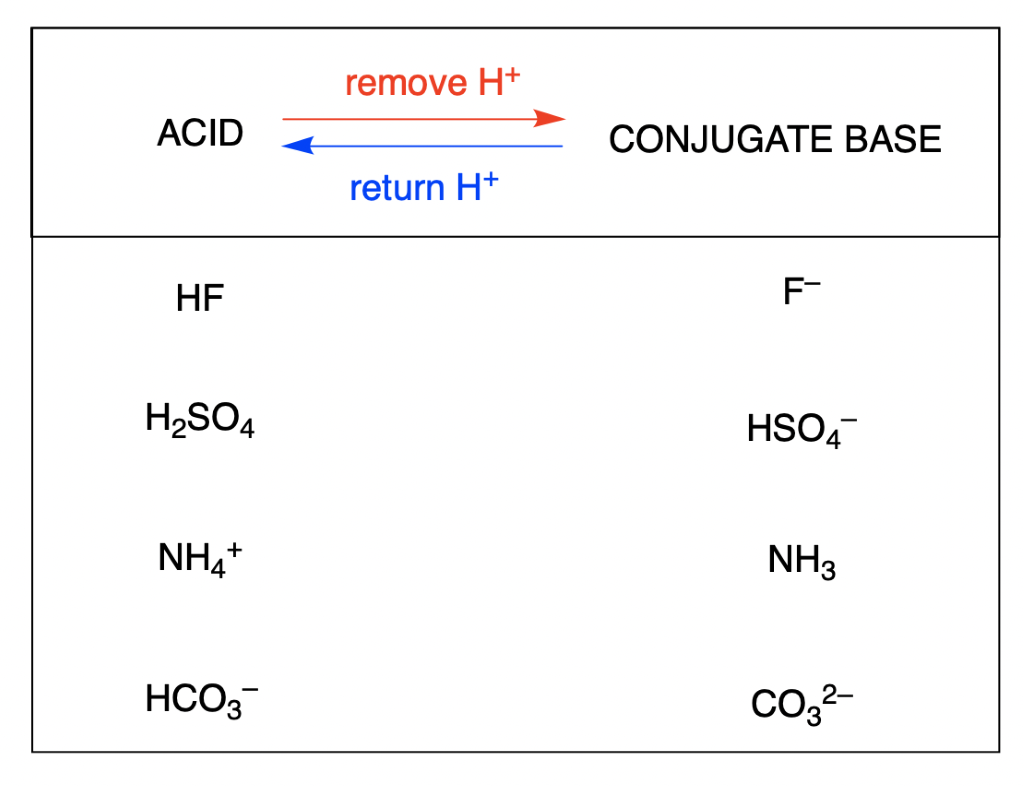
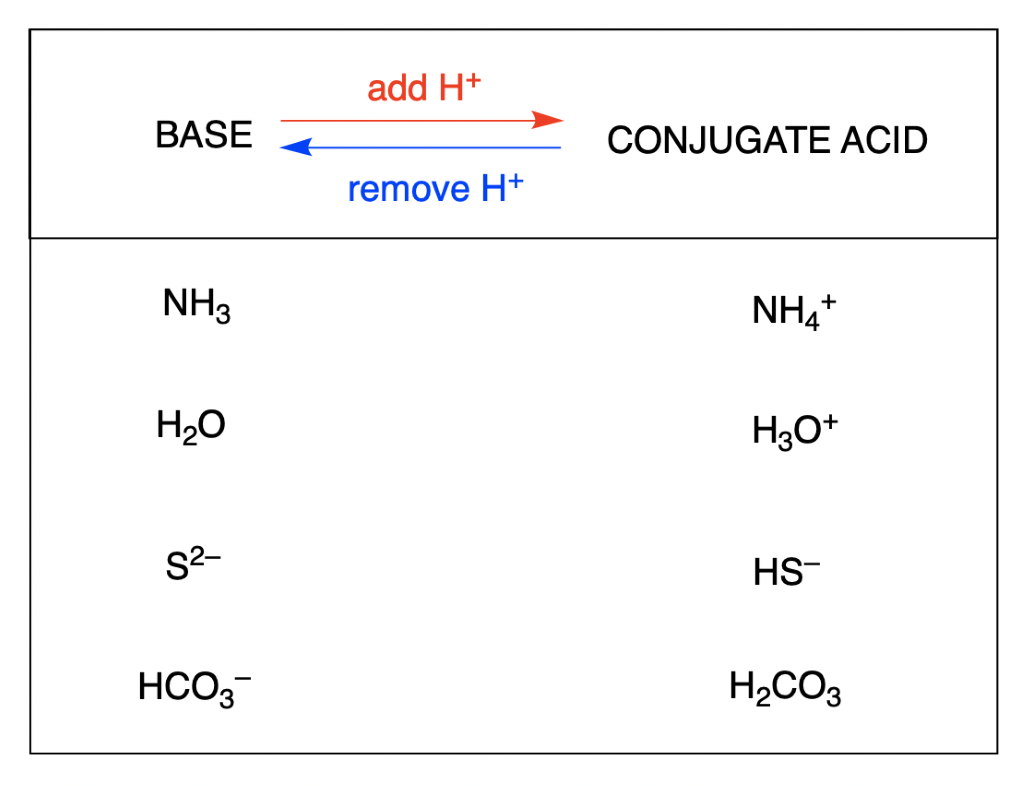
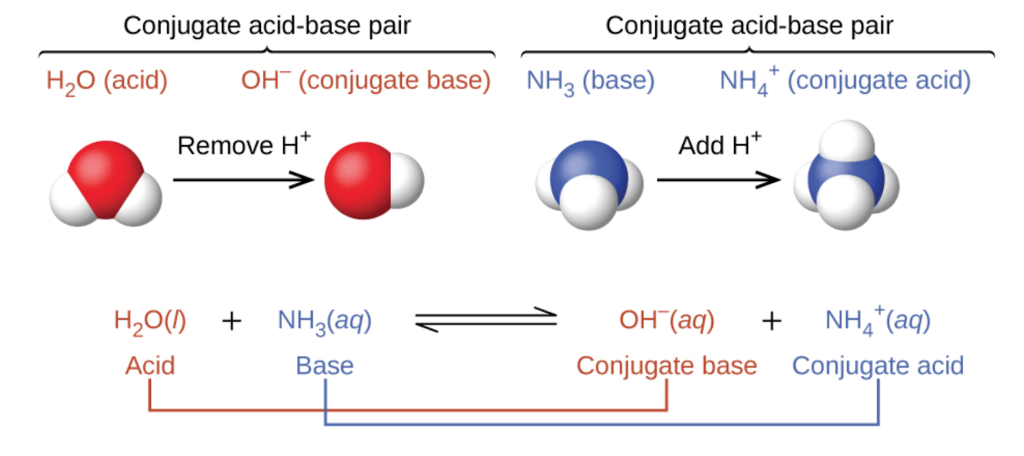
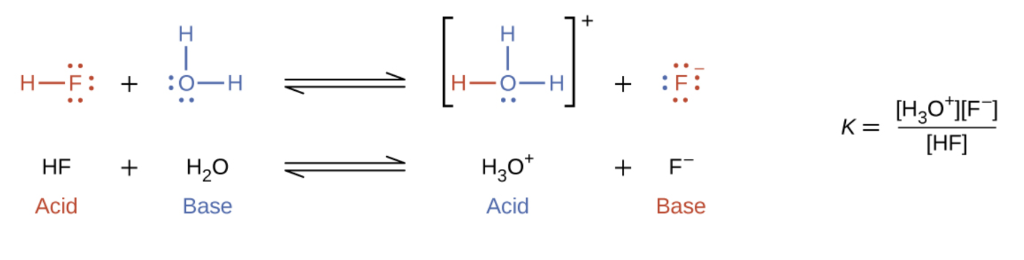
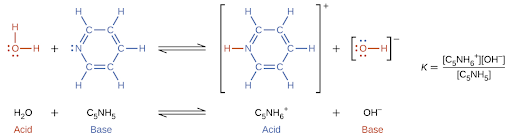
 Toutes les
Toutes les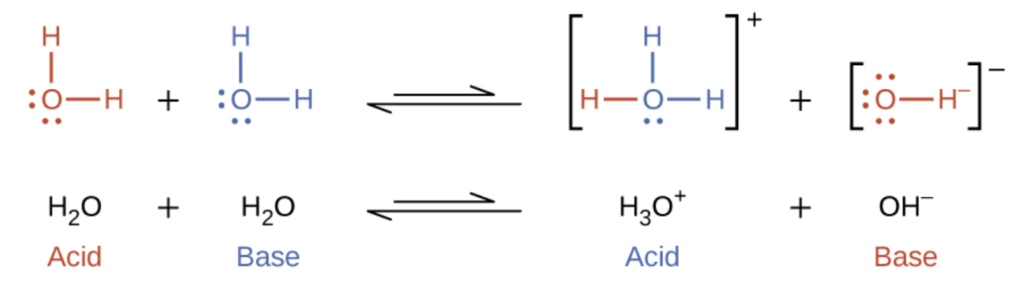
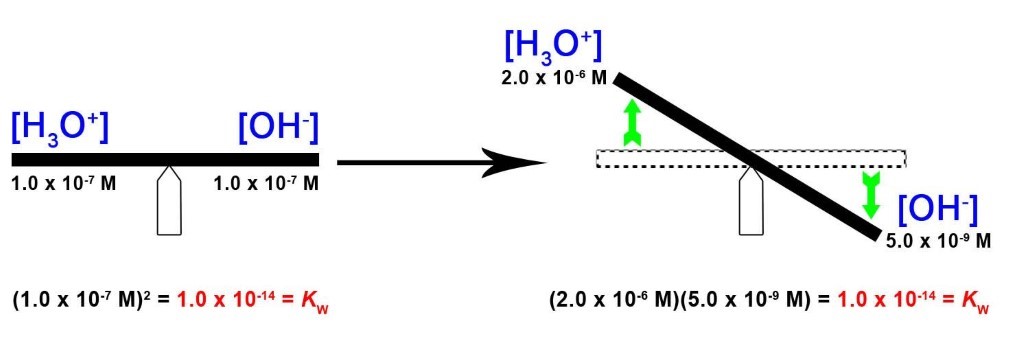
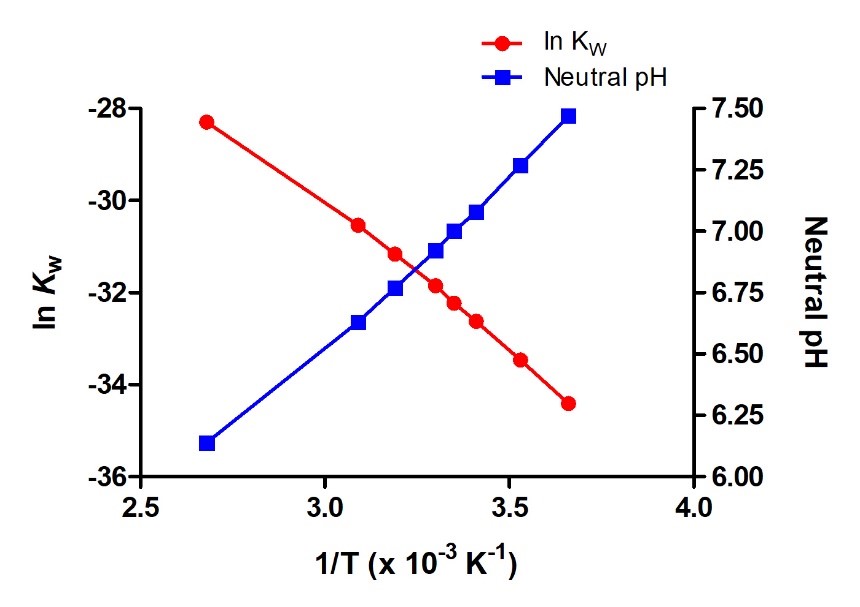
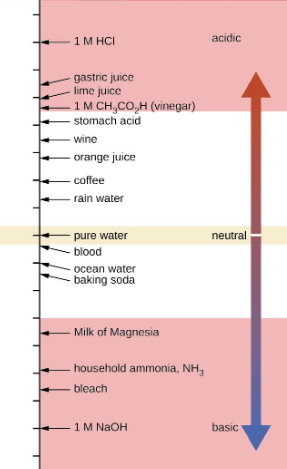



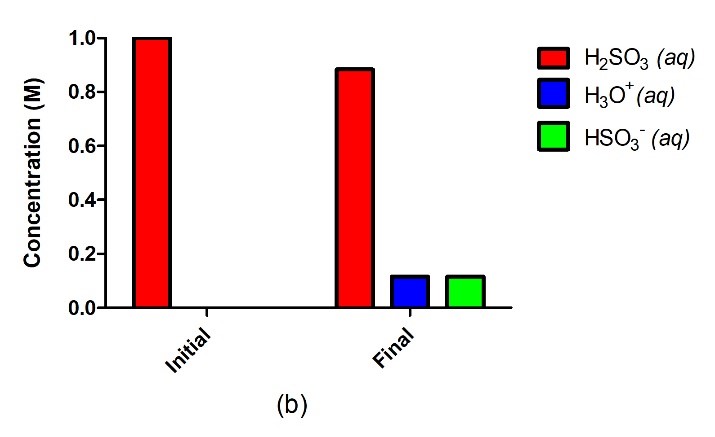
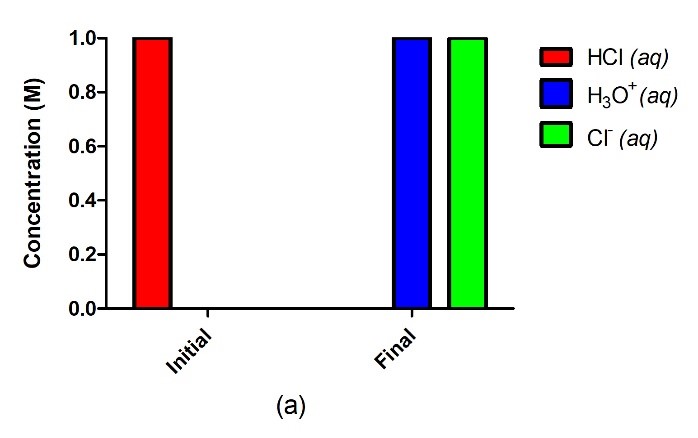
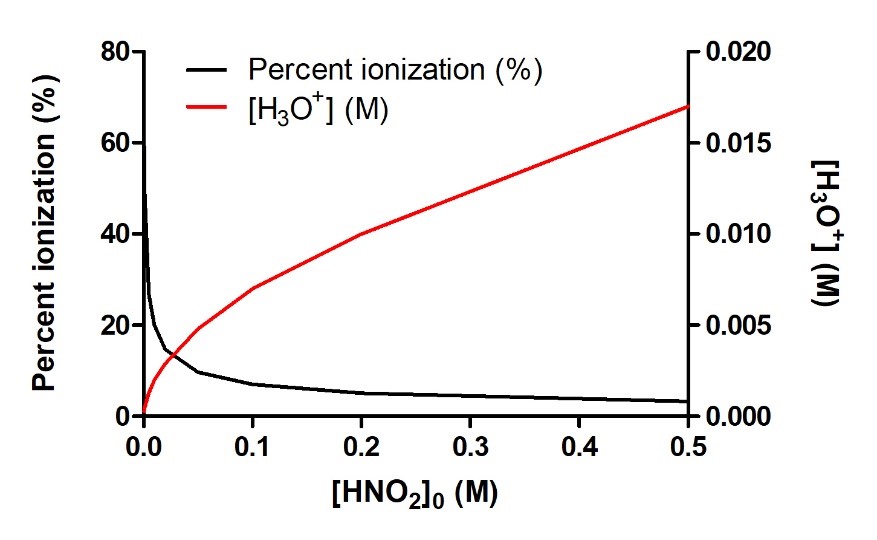
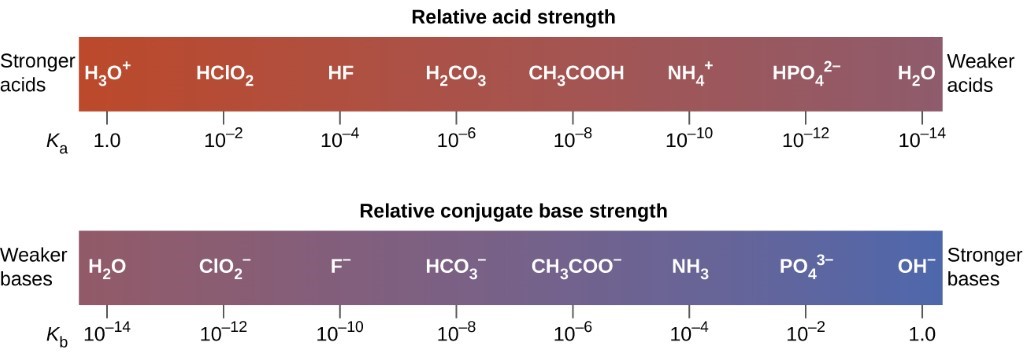
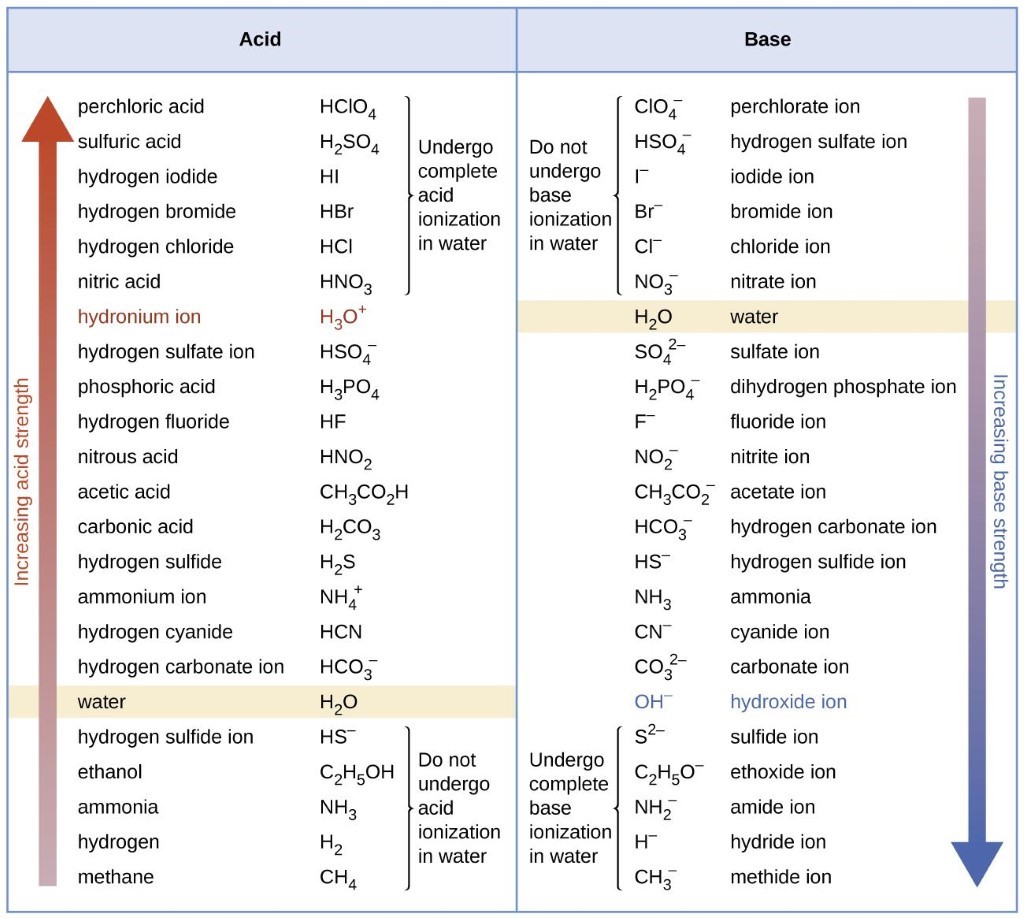



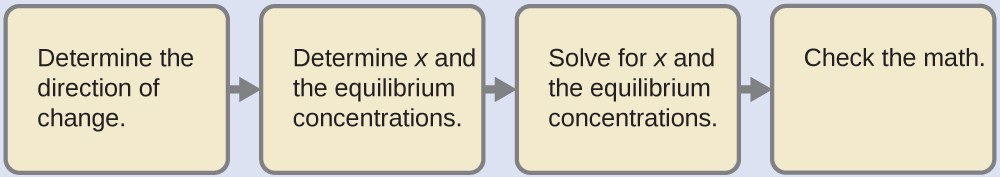

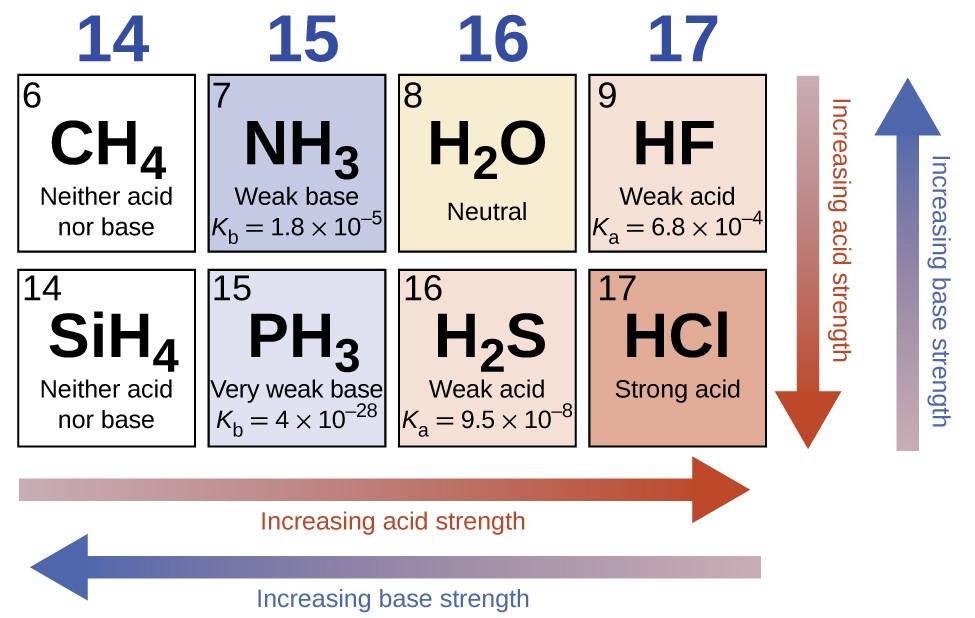
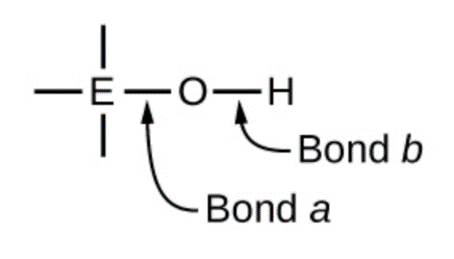
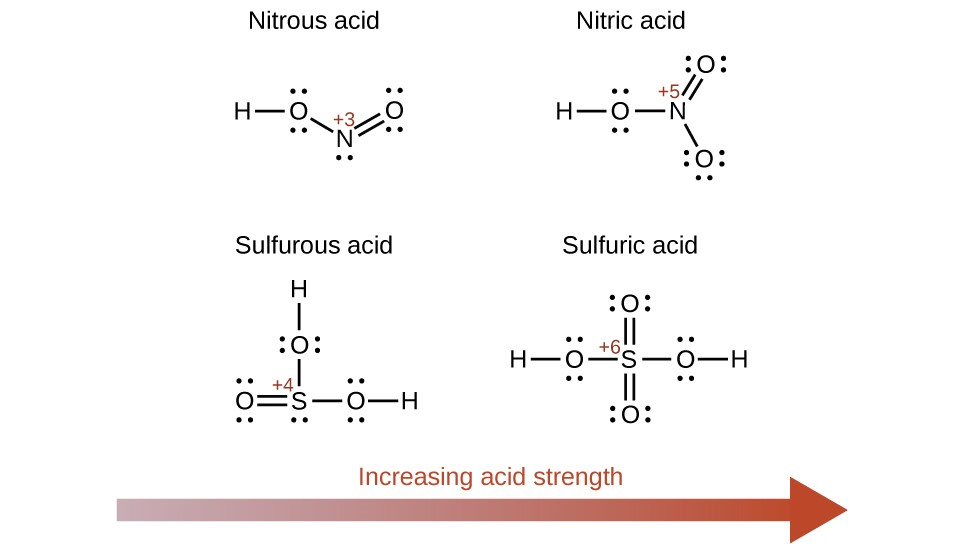

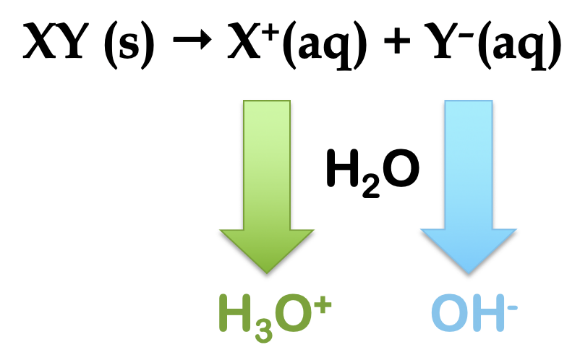
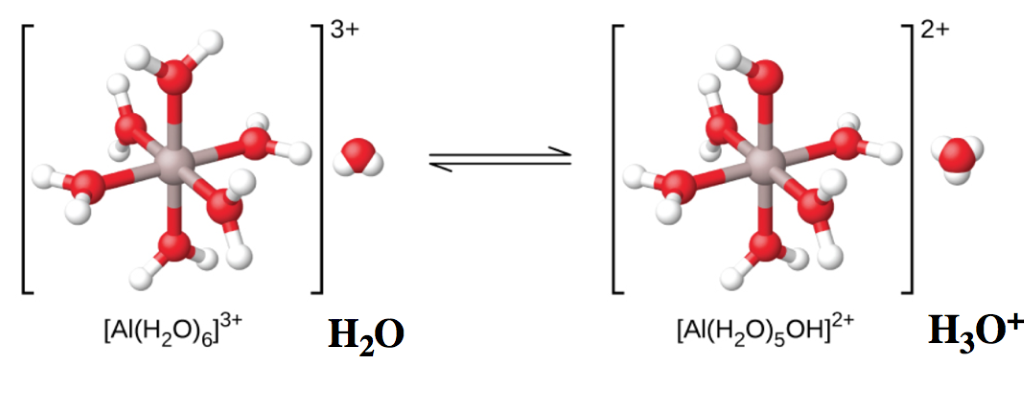

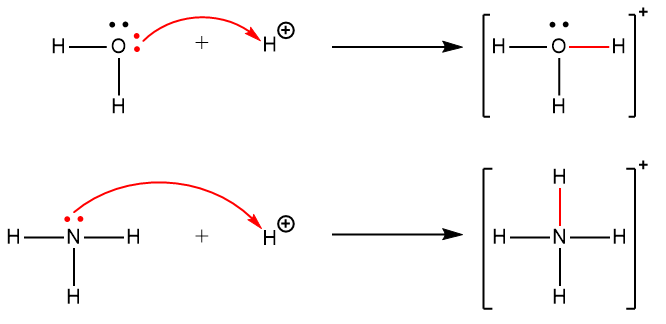
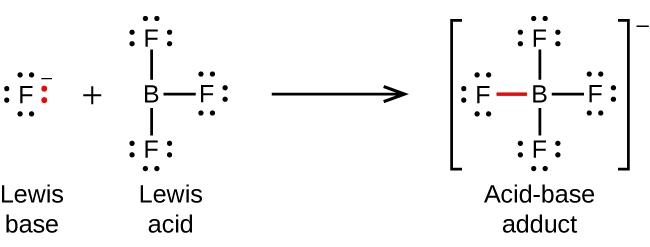
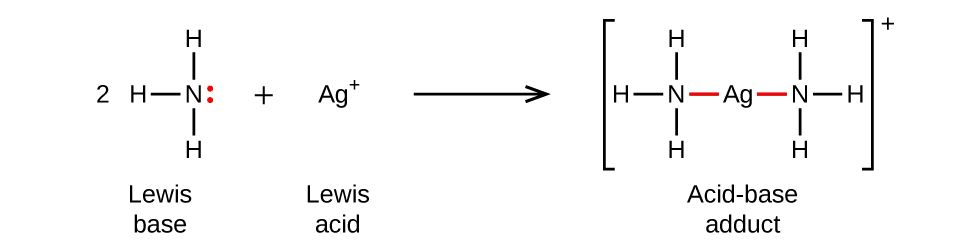
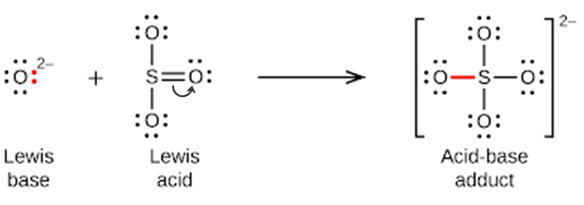
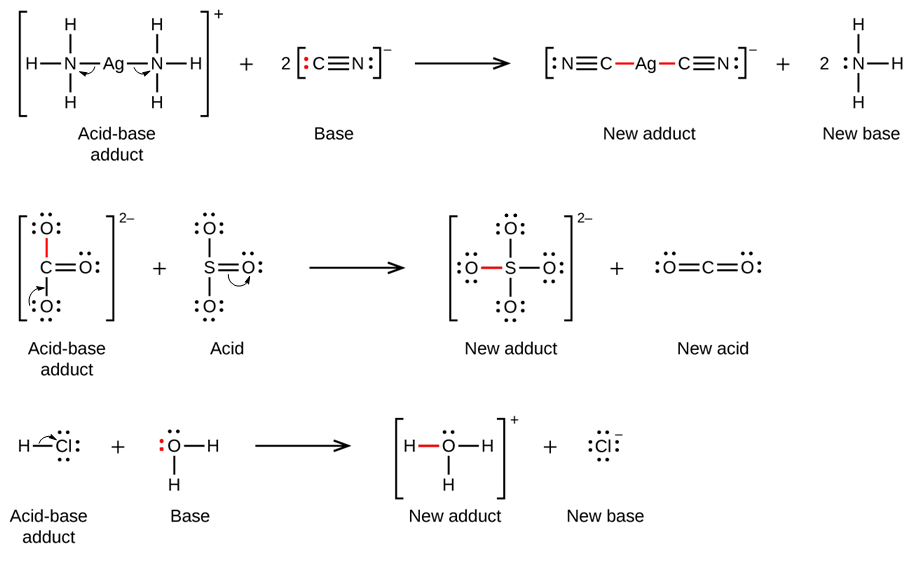
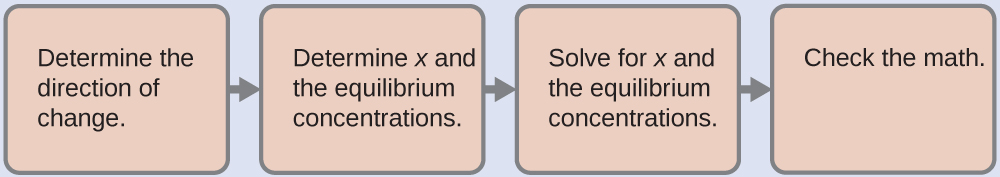
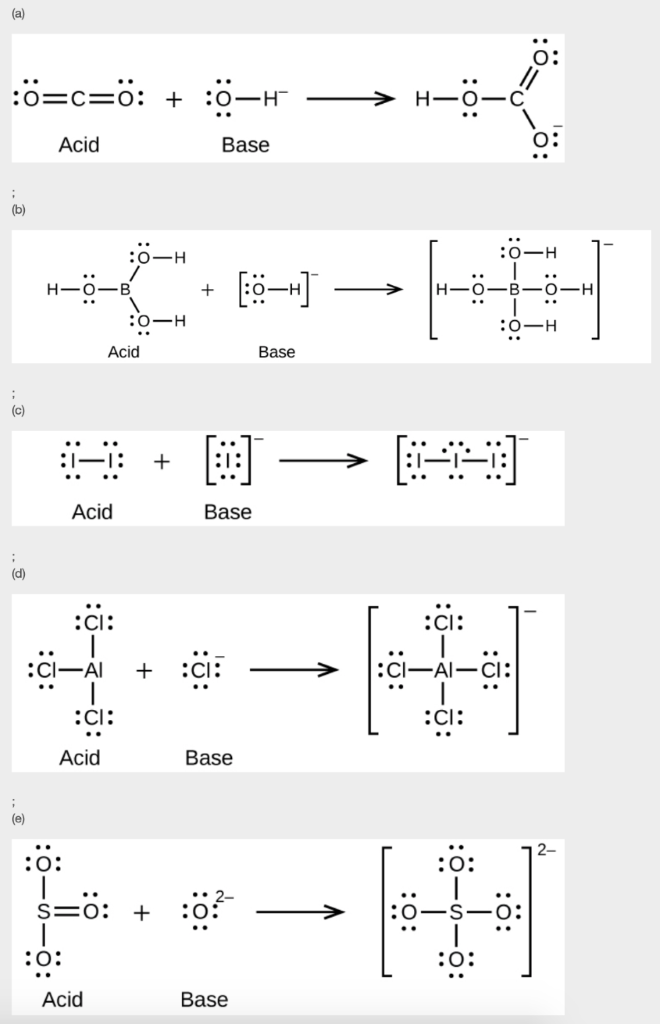
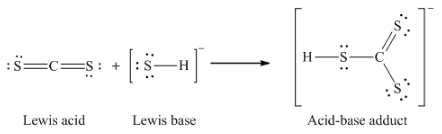
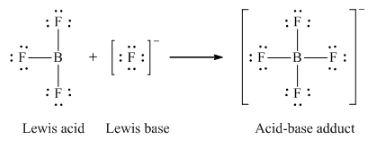
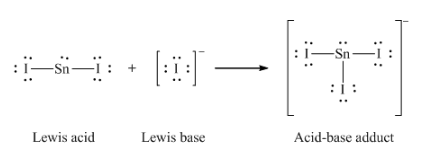
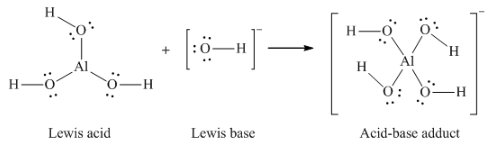
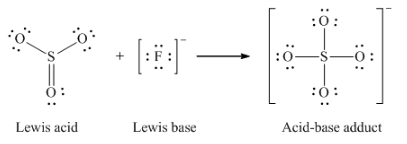
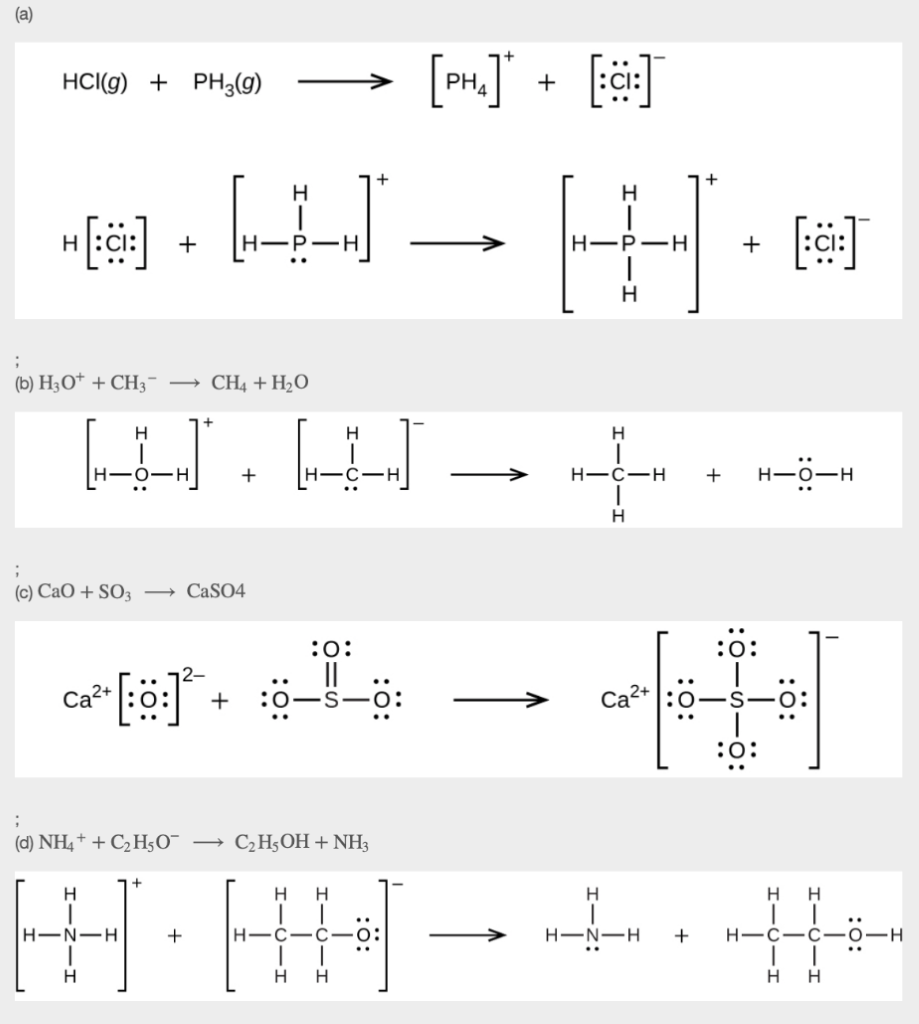
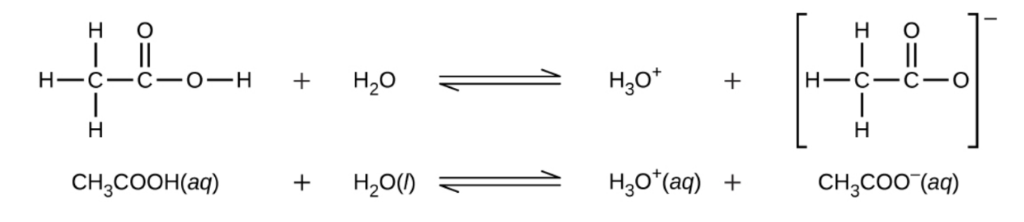
 NOTE : Les
NOTE : Les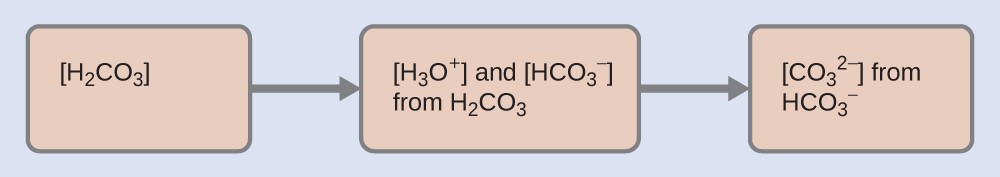
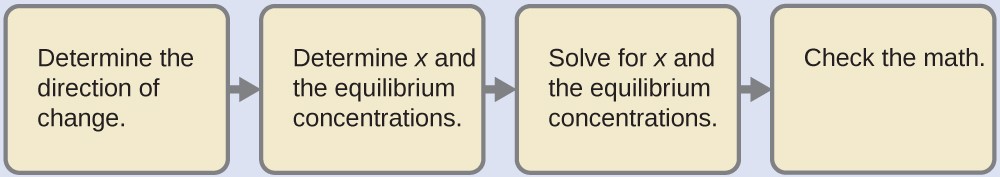
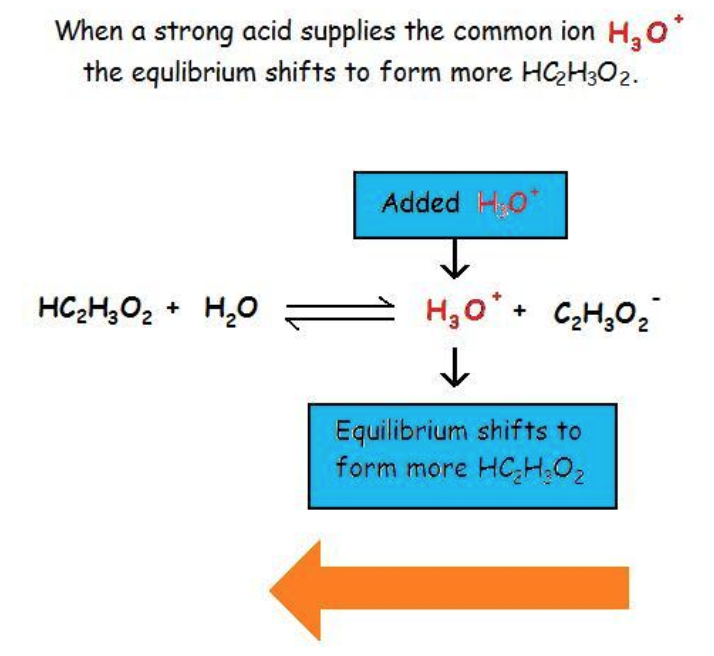
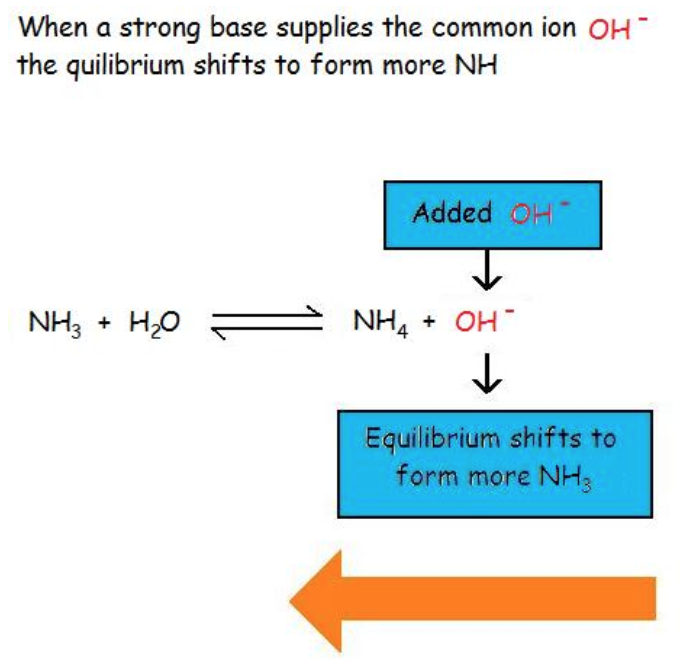

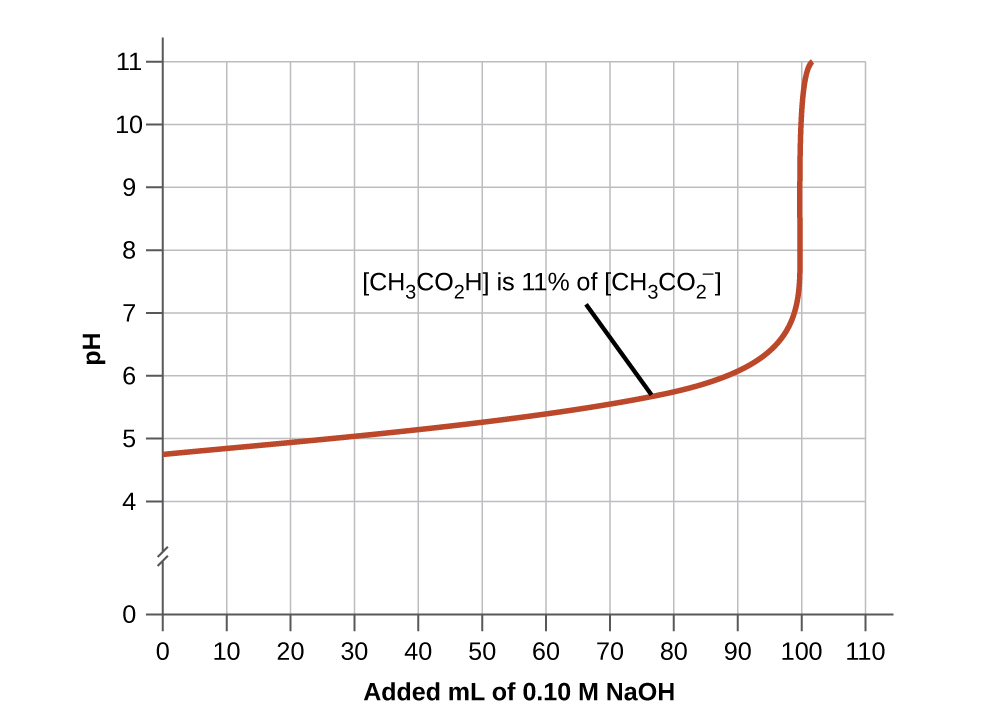

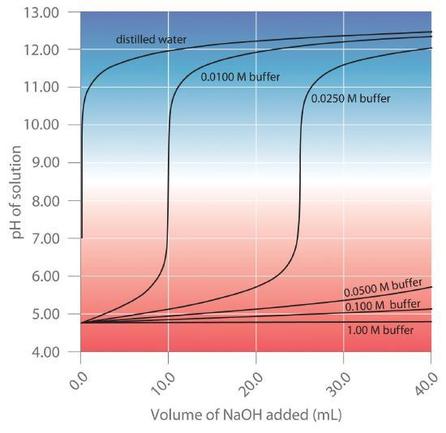


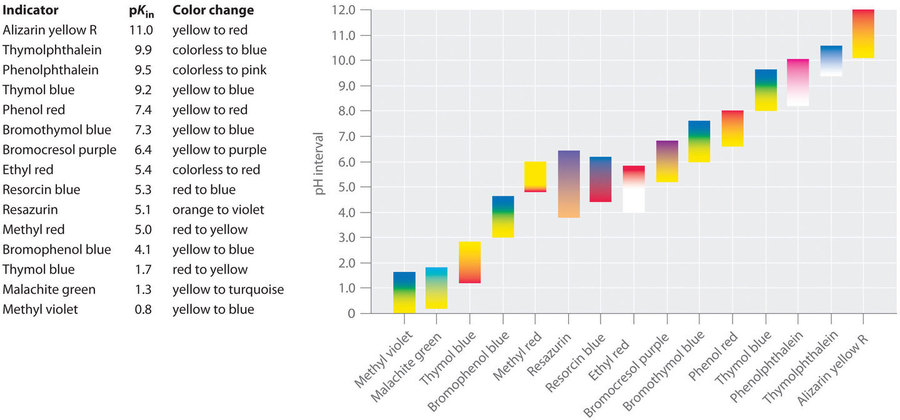
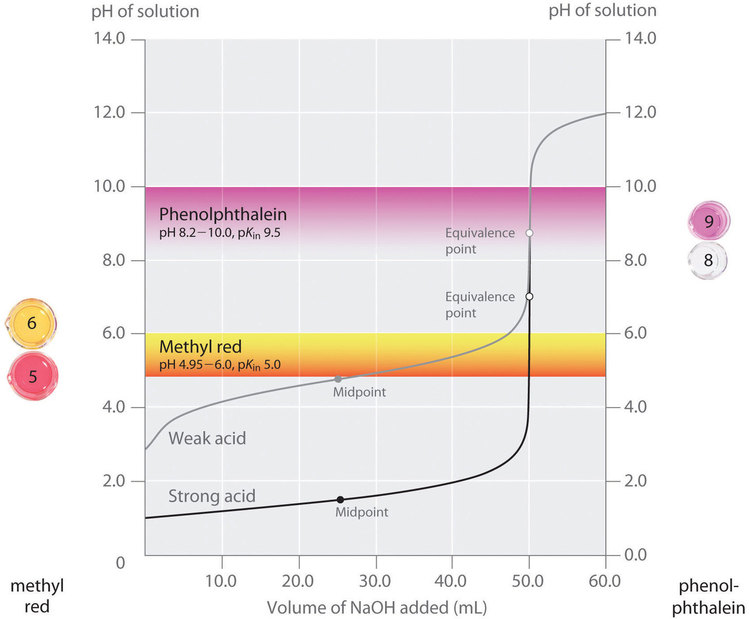

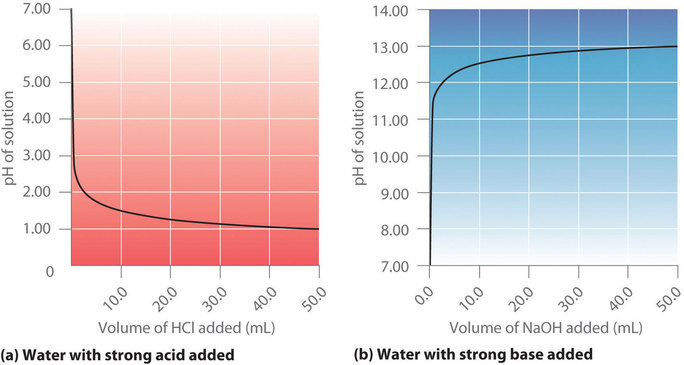
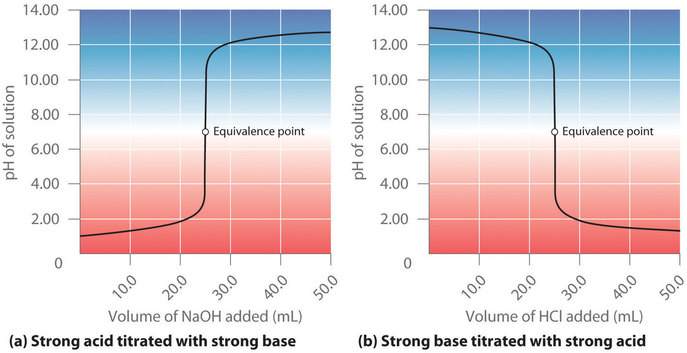
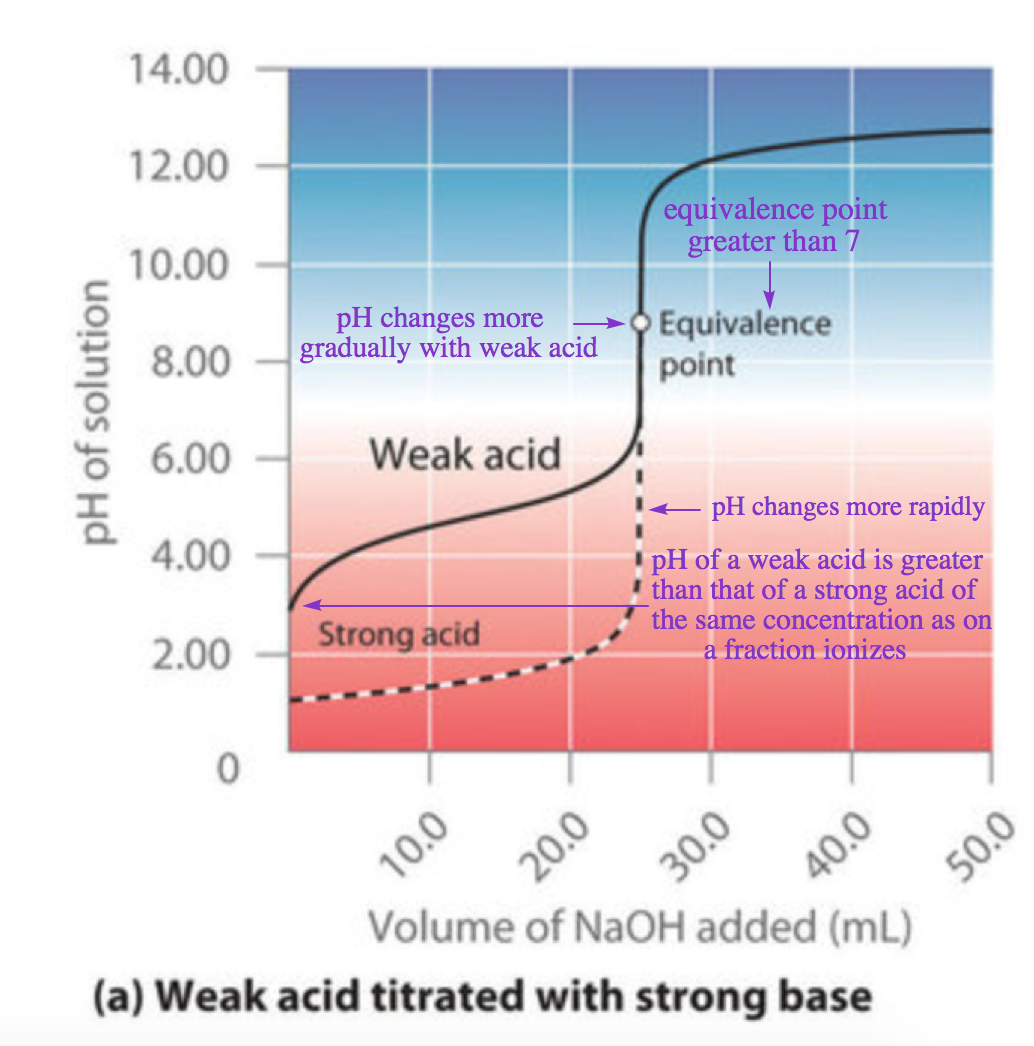
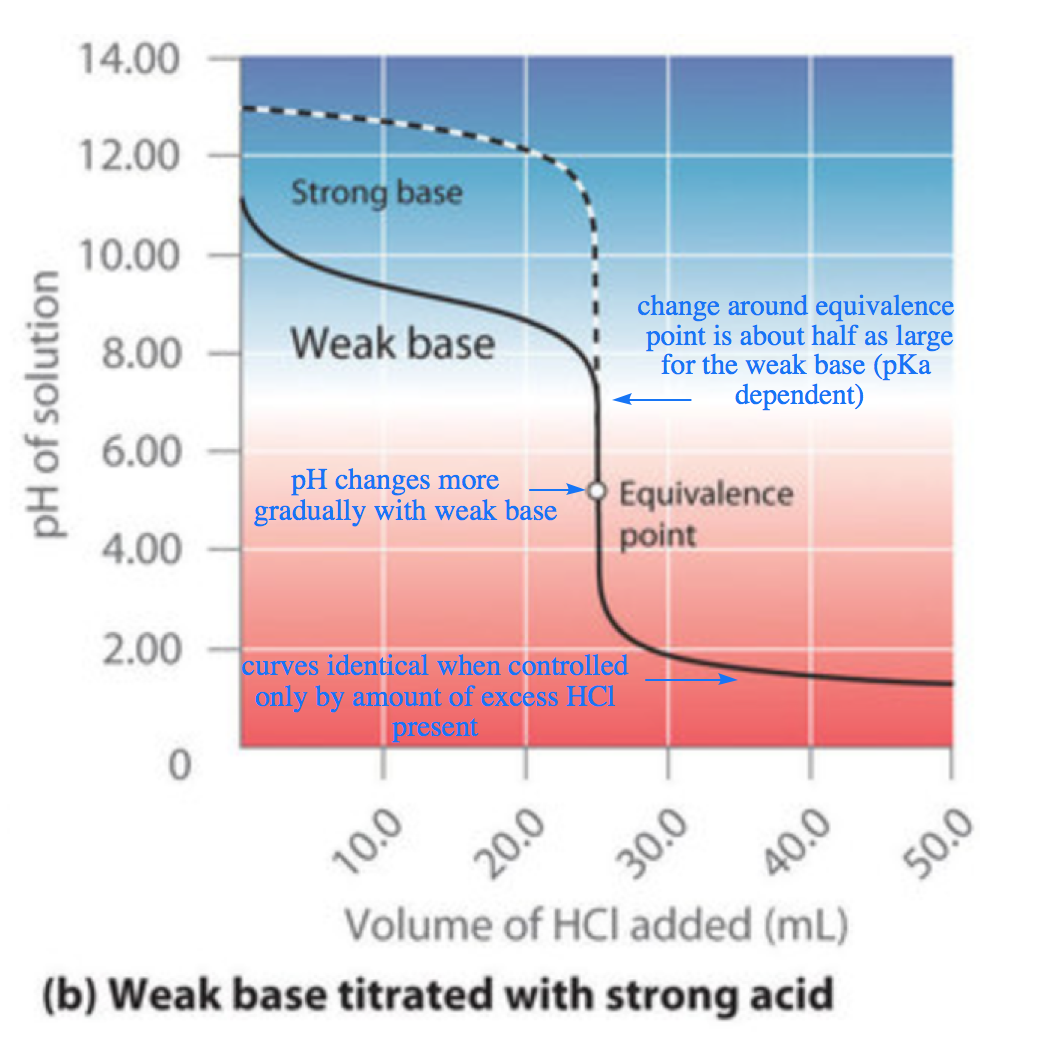
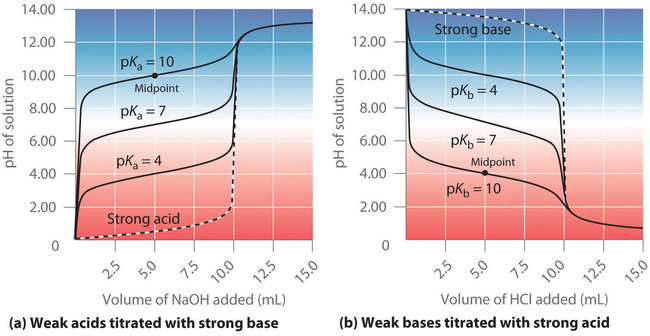
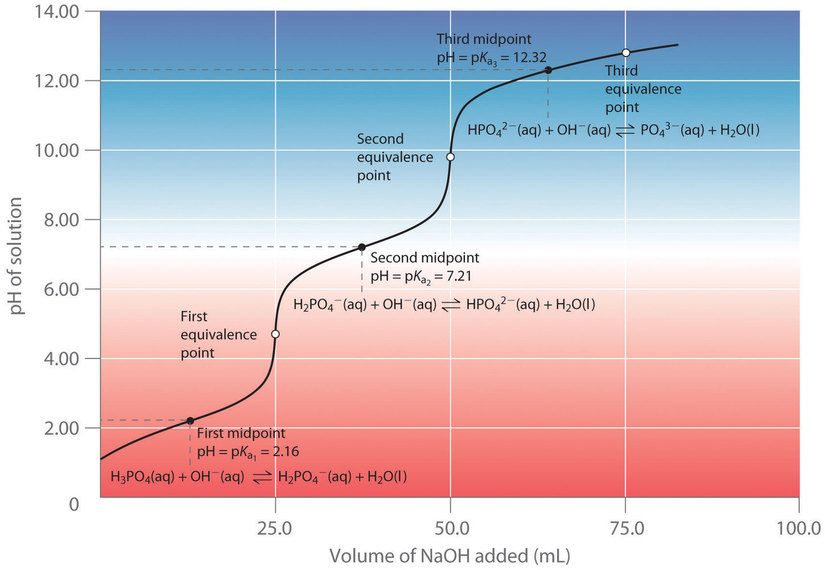

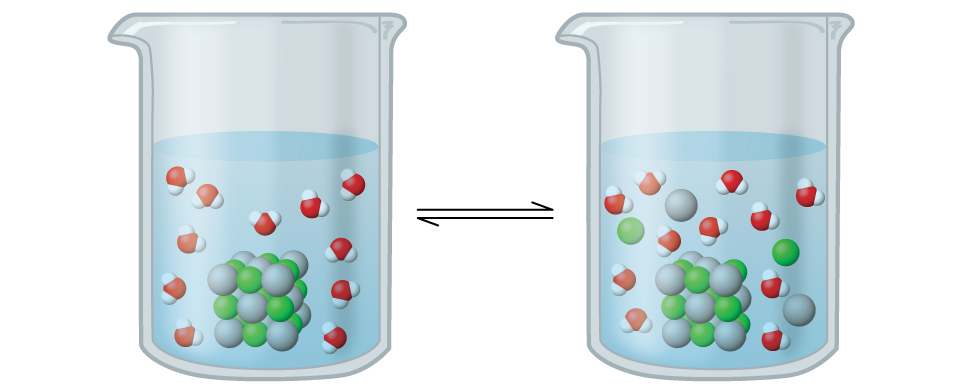
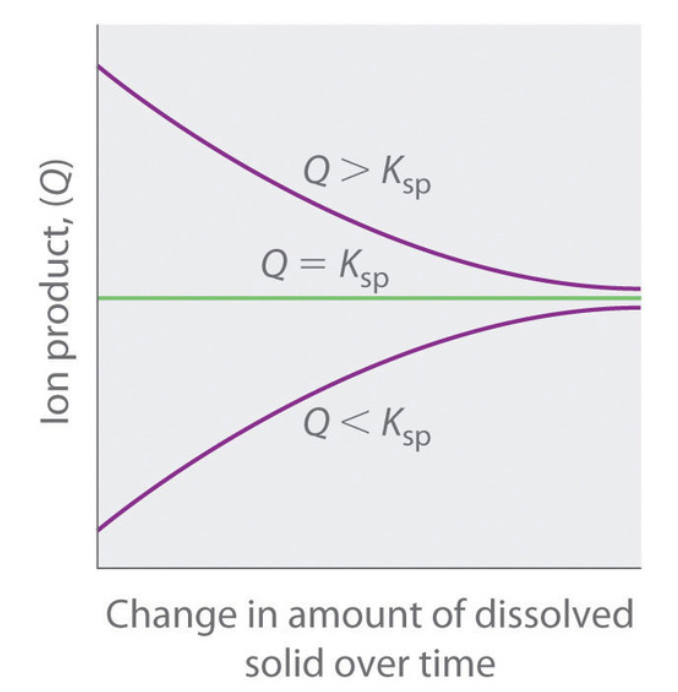

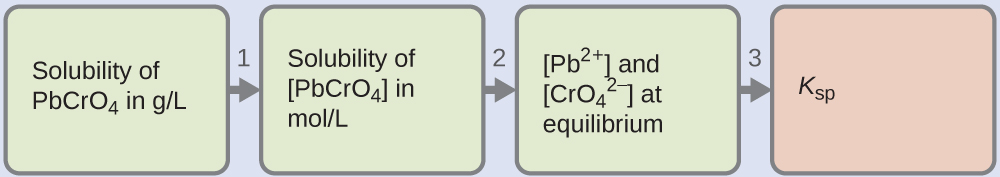
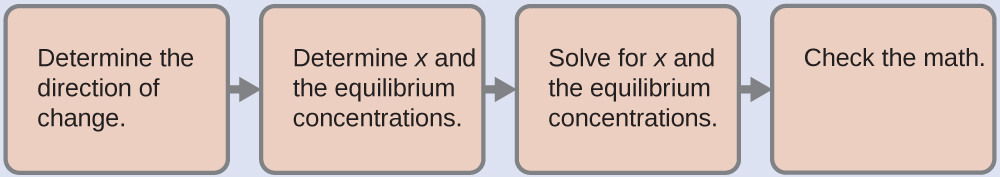



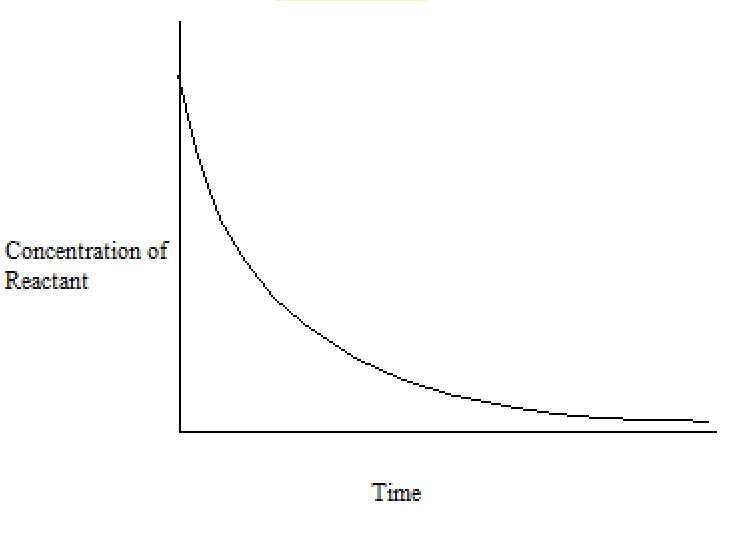
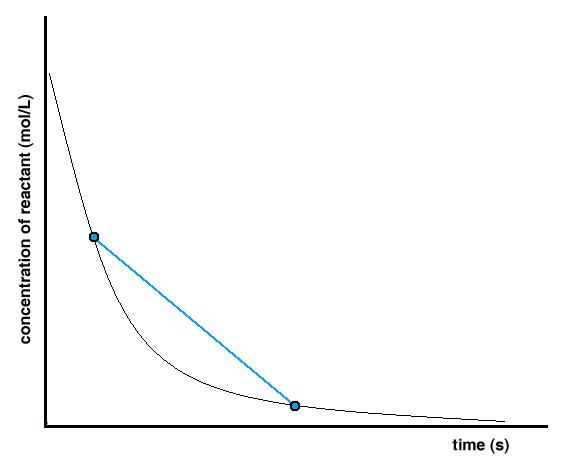
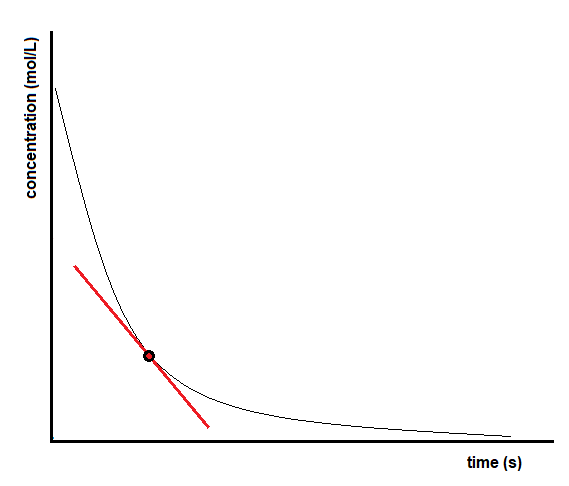
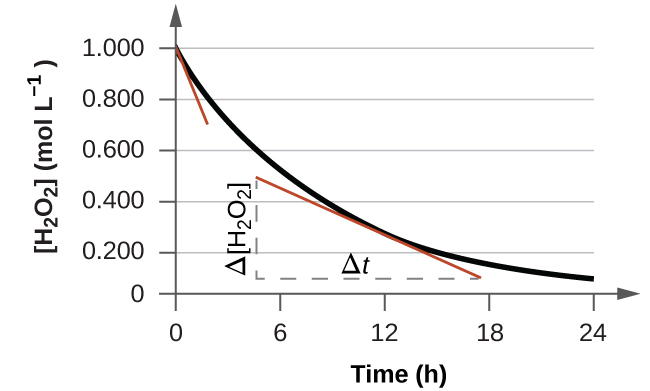
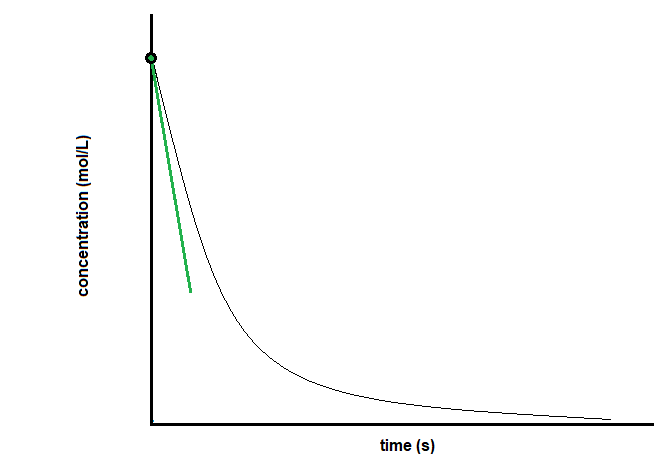

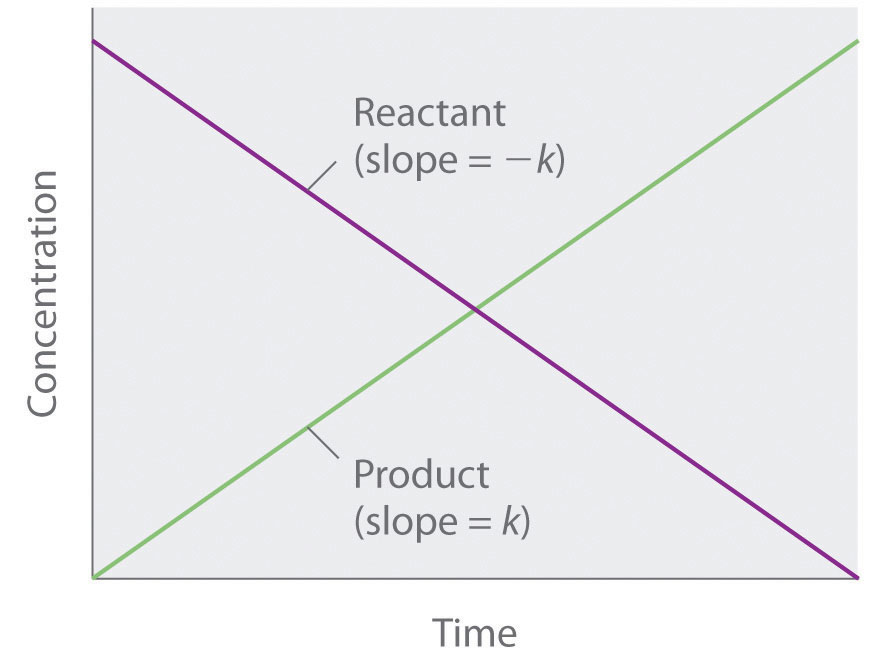
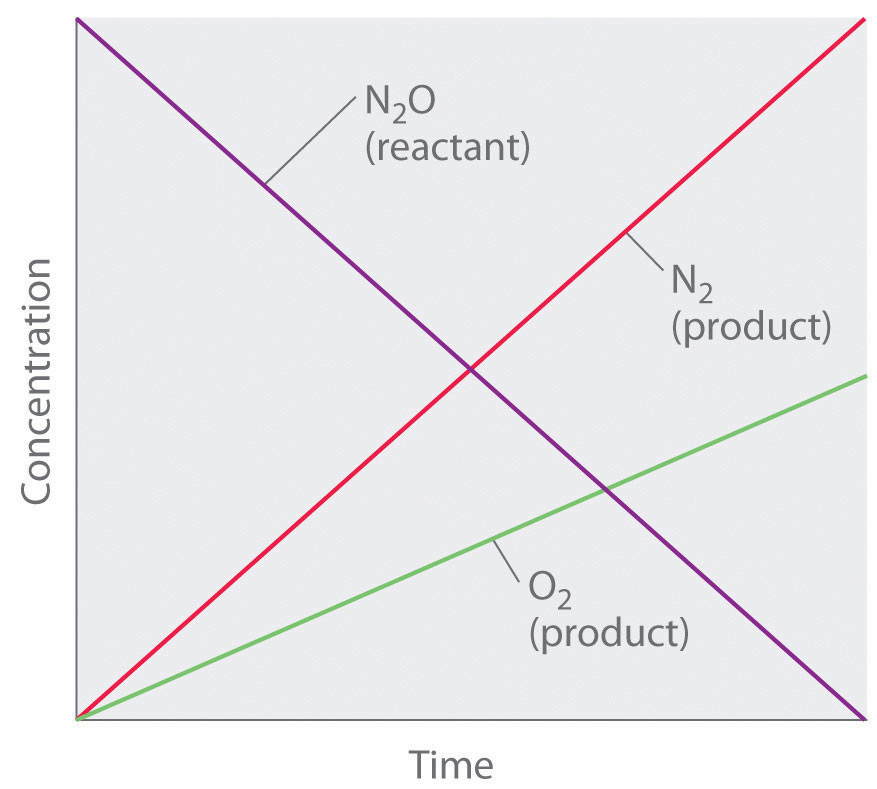
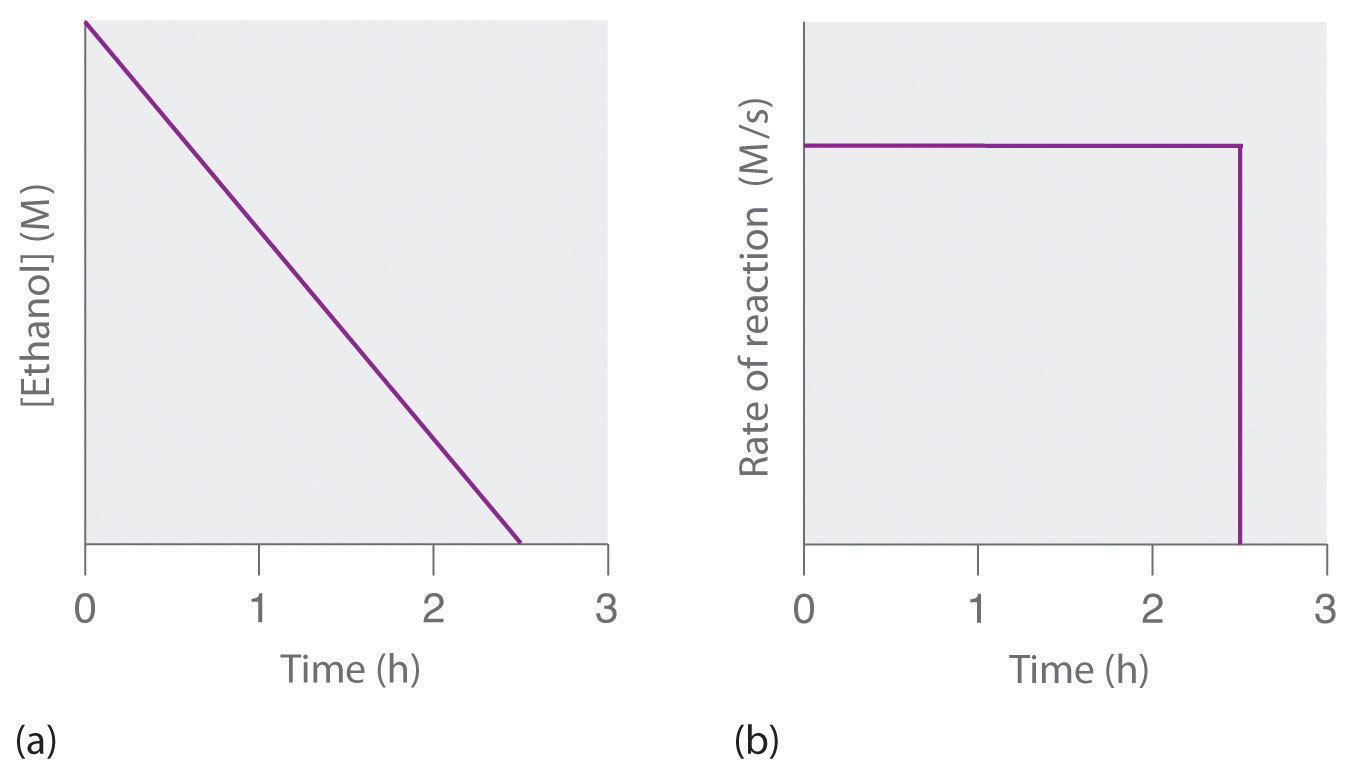
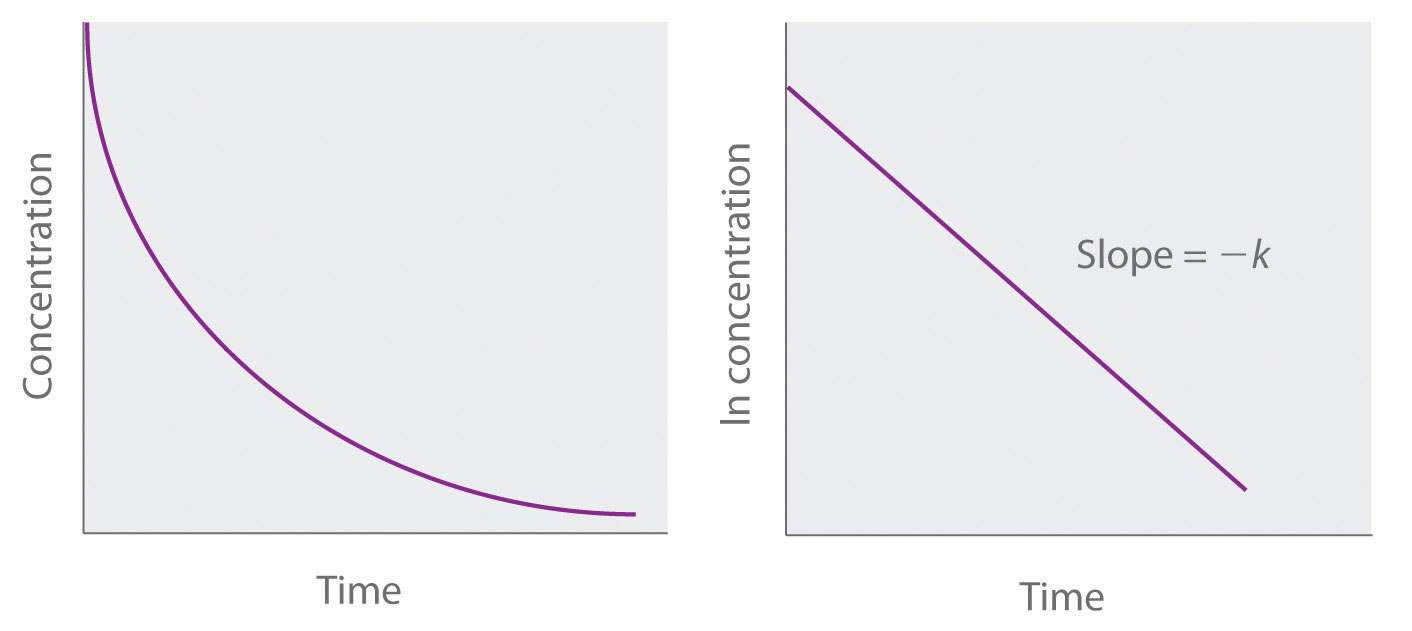
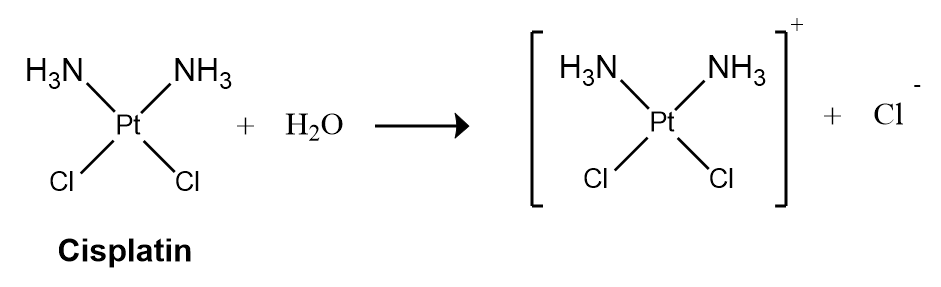
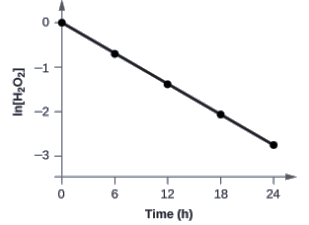
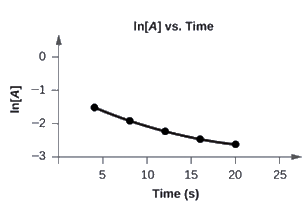
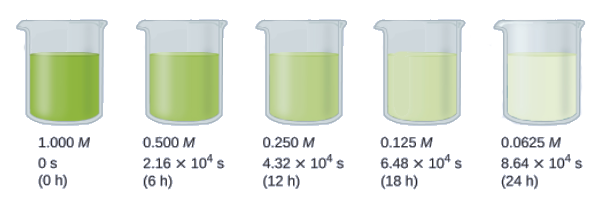
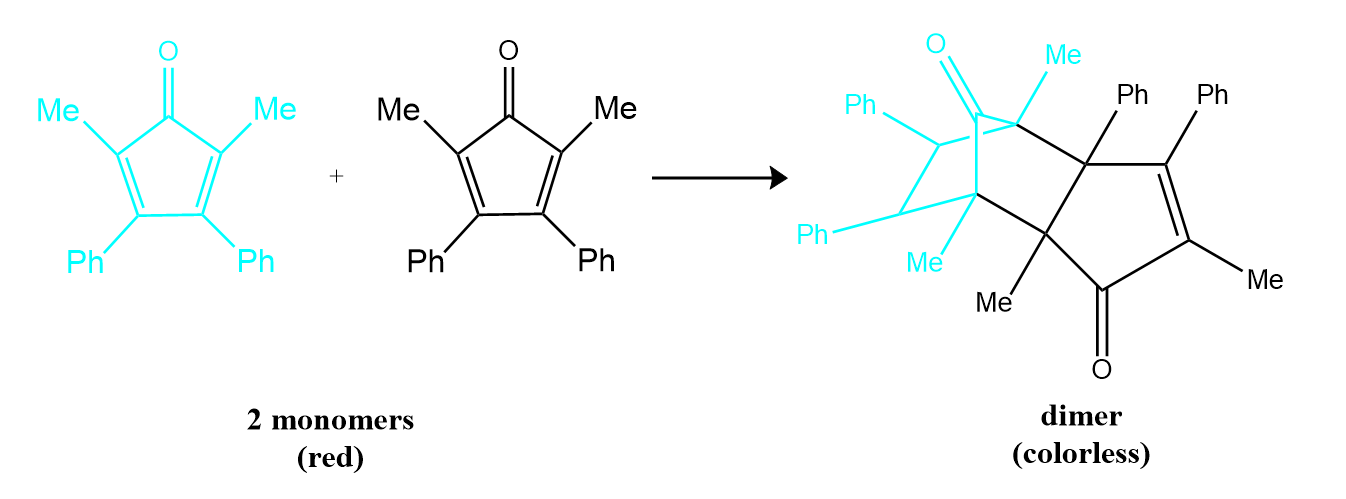
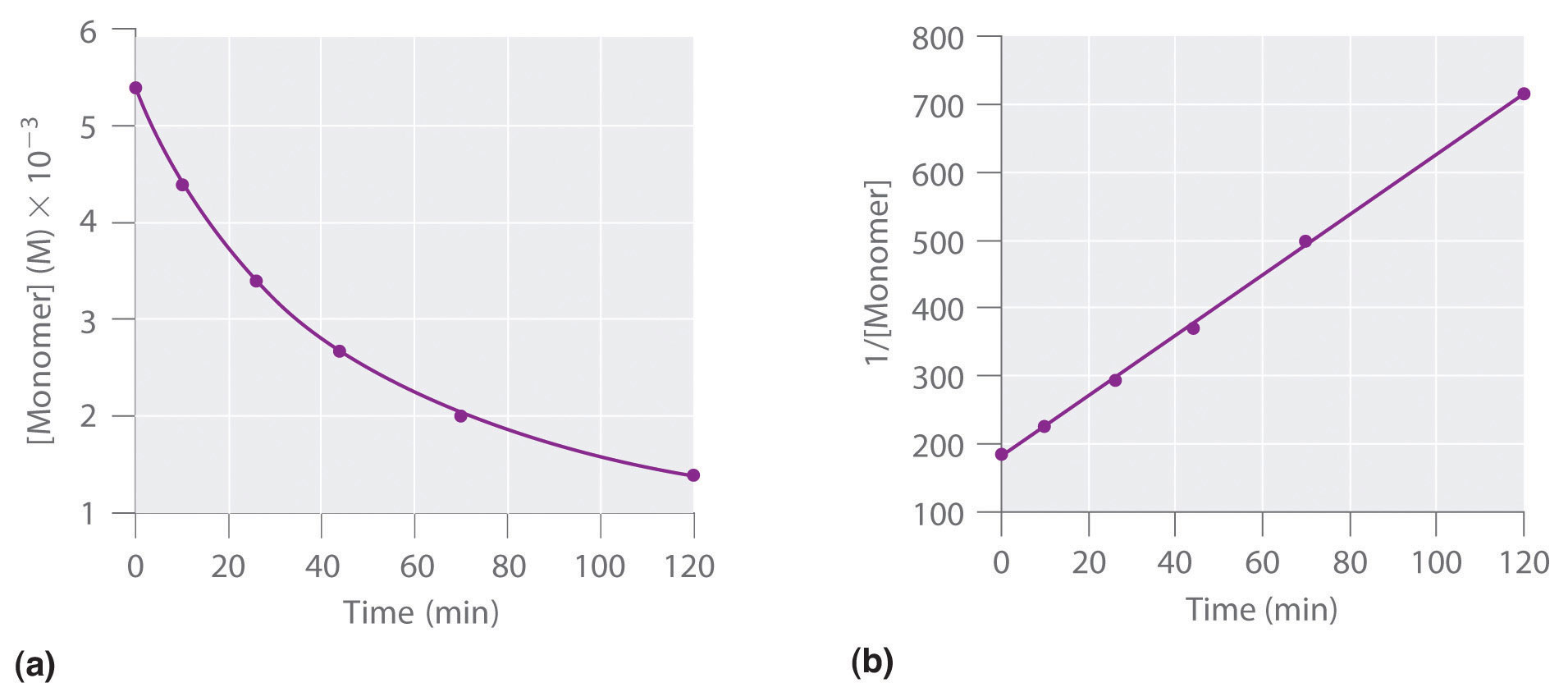
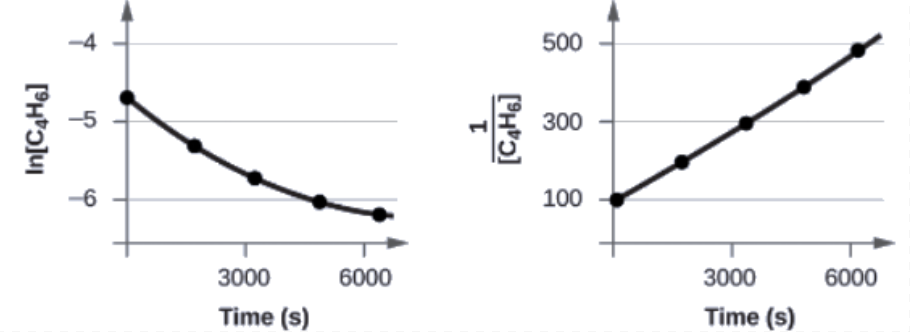
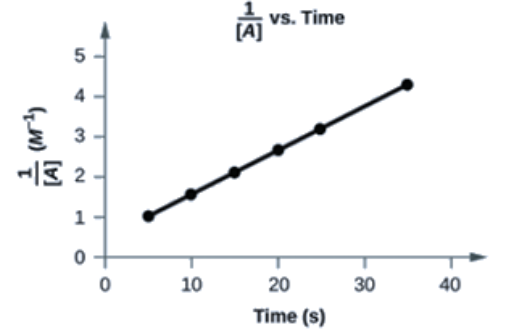
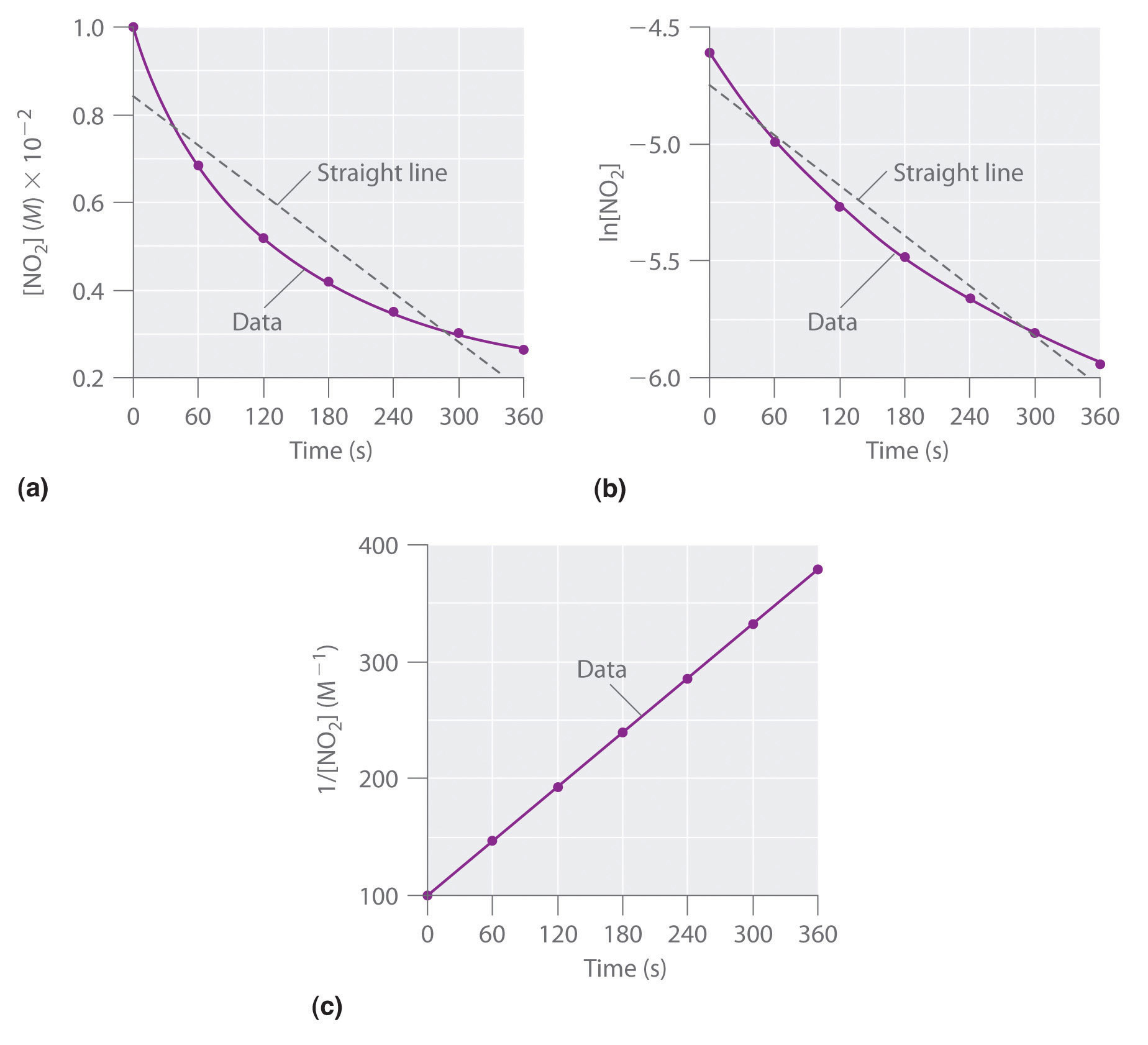
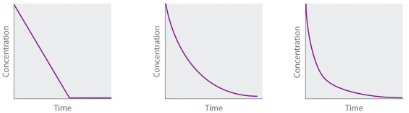
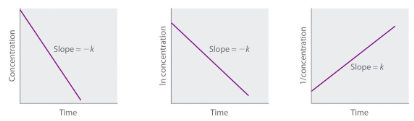
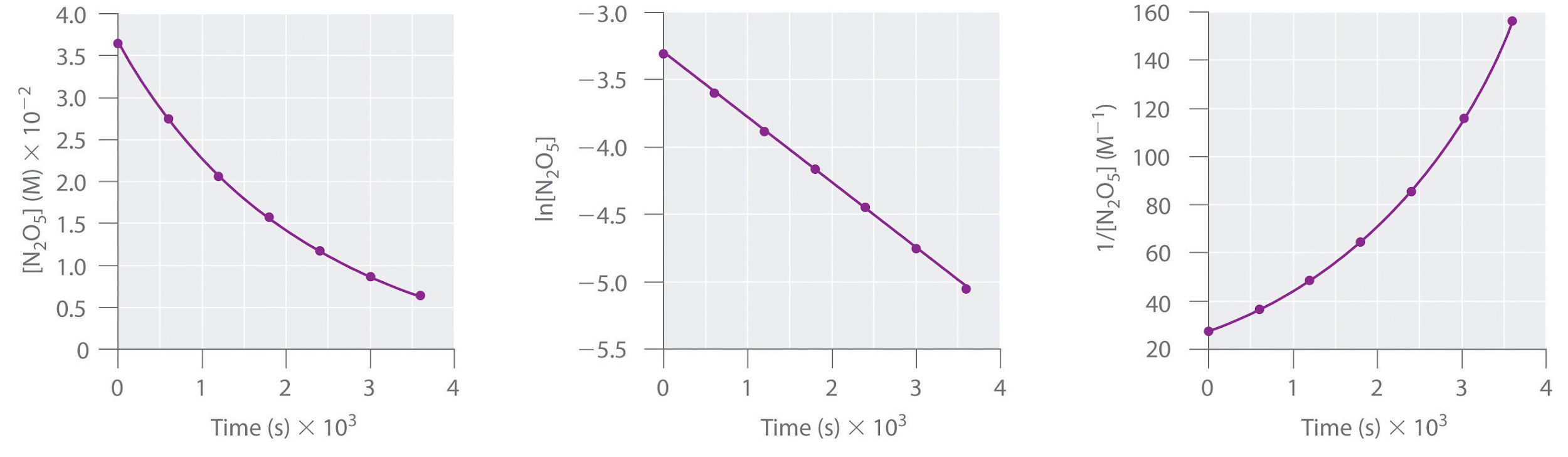
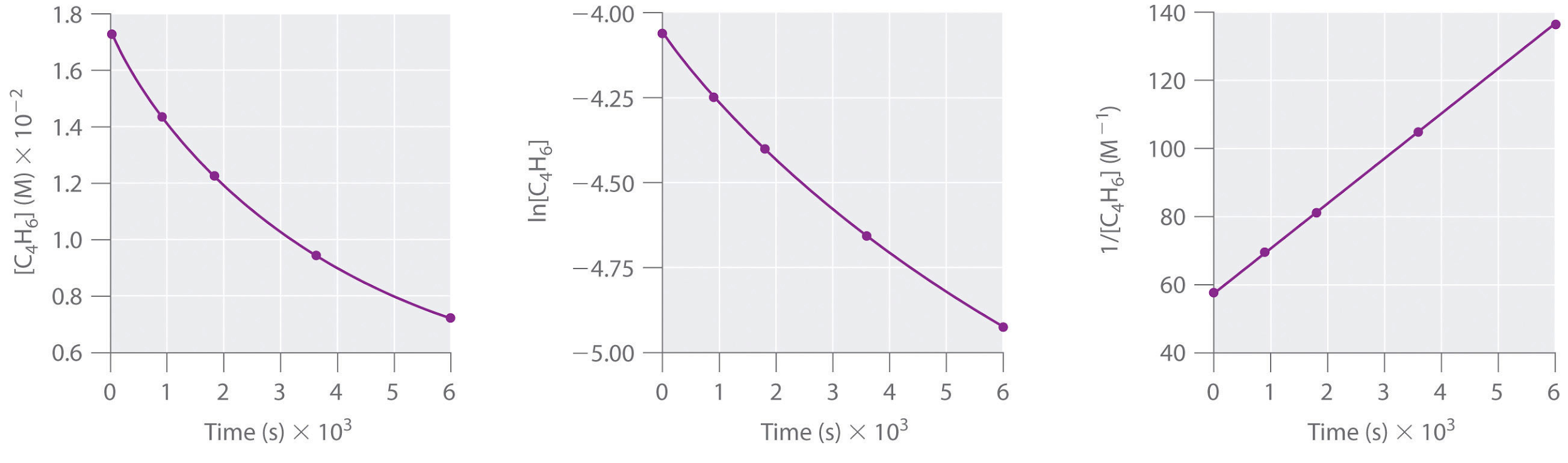
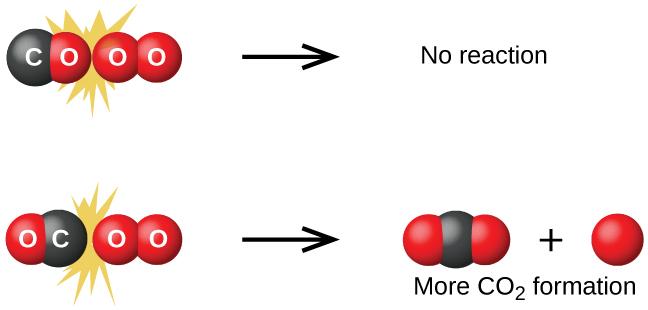
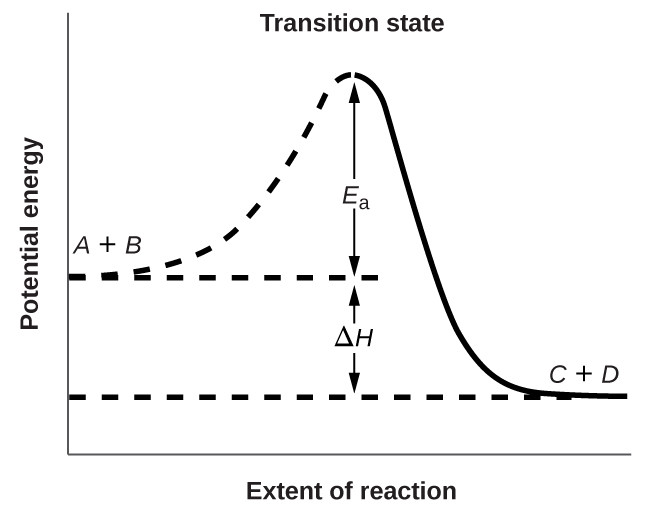
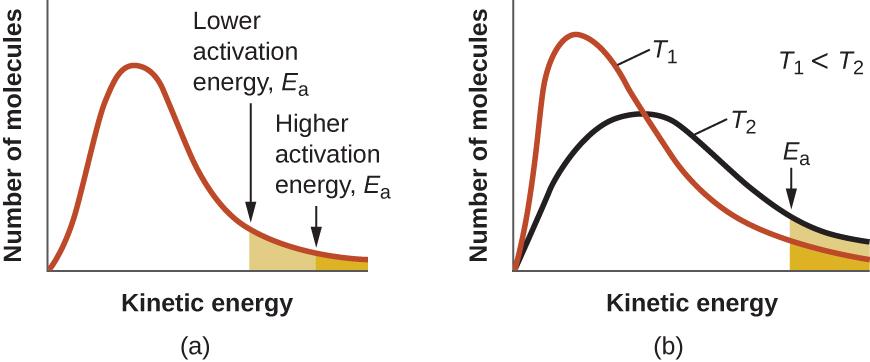
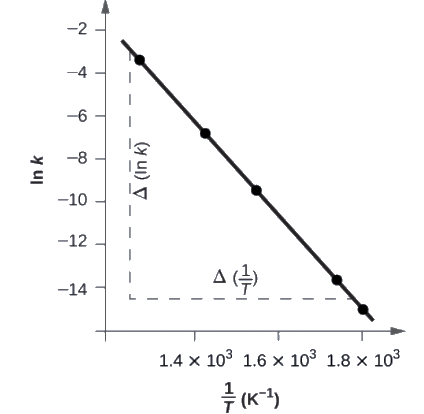
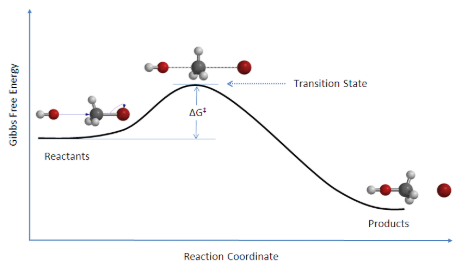
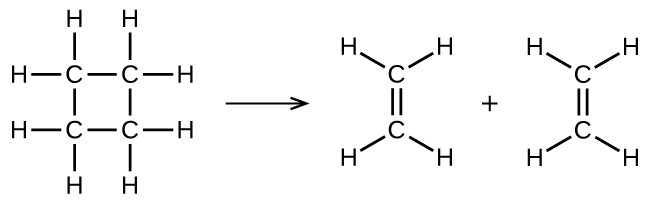
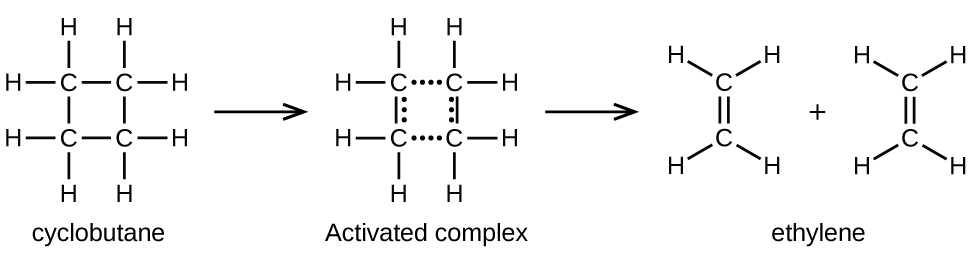
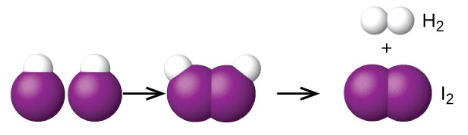

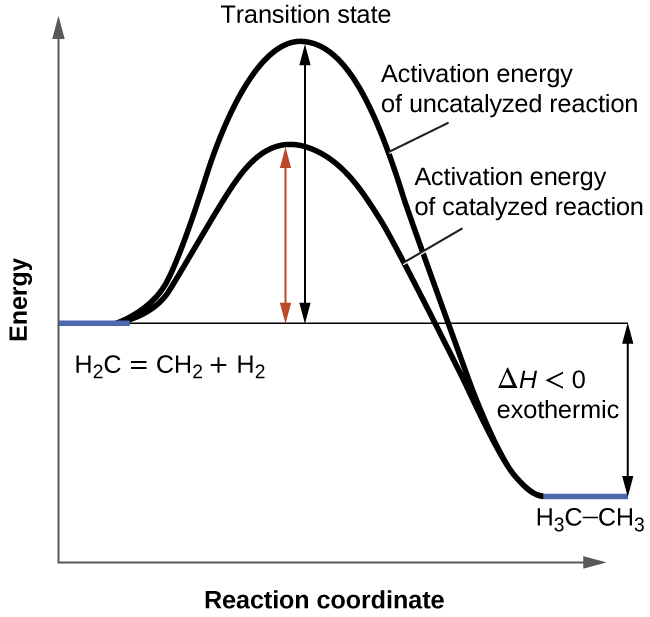
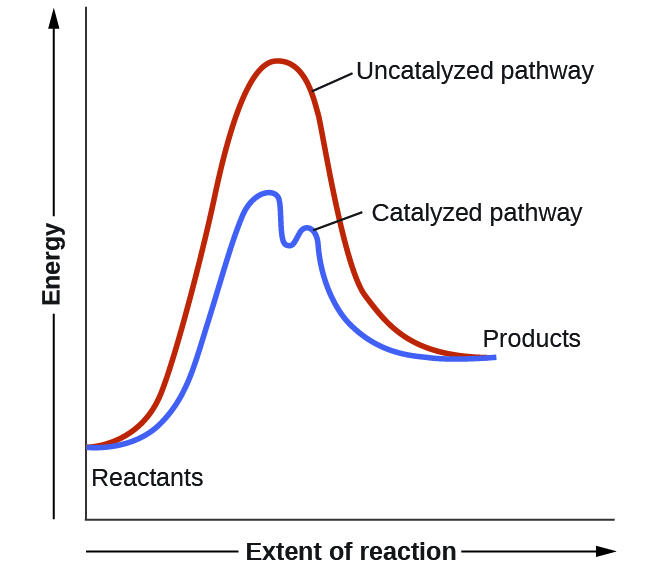
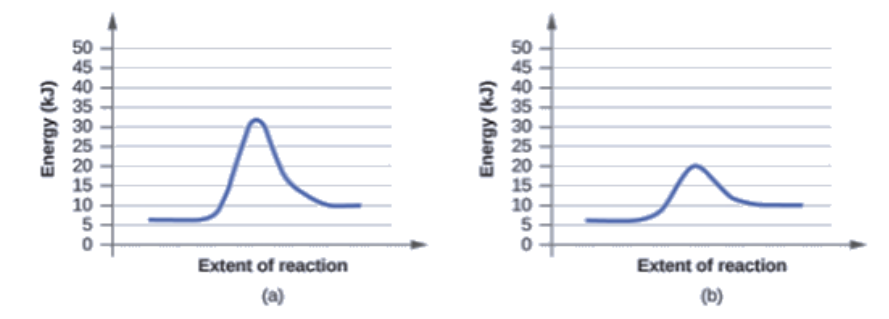
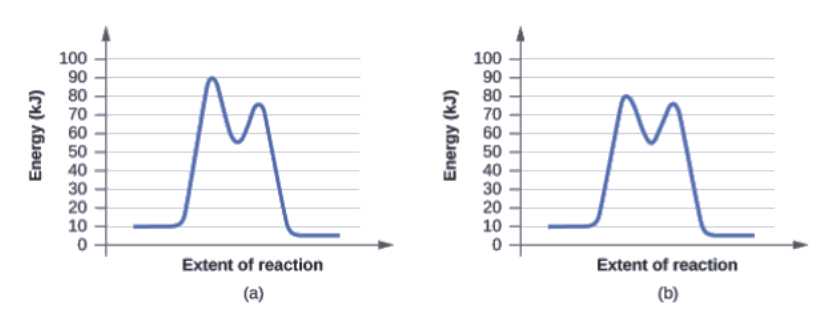
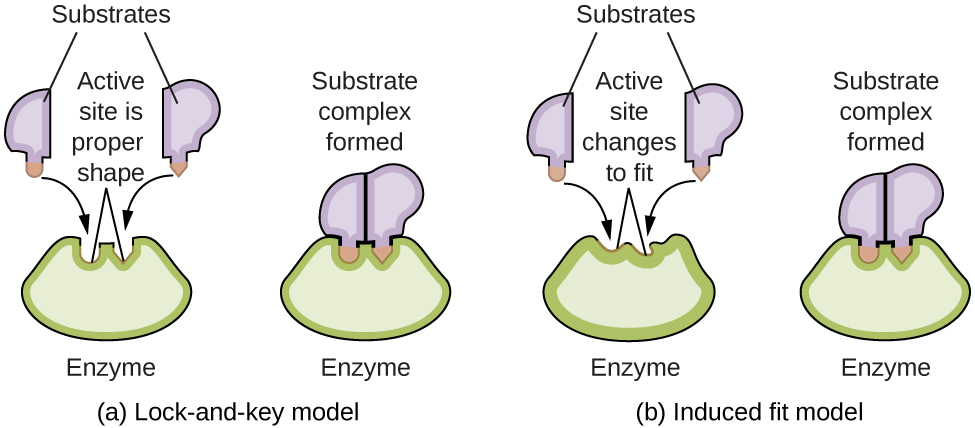





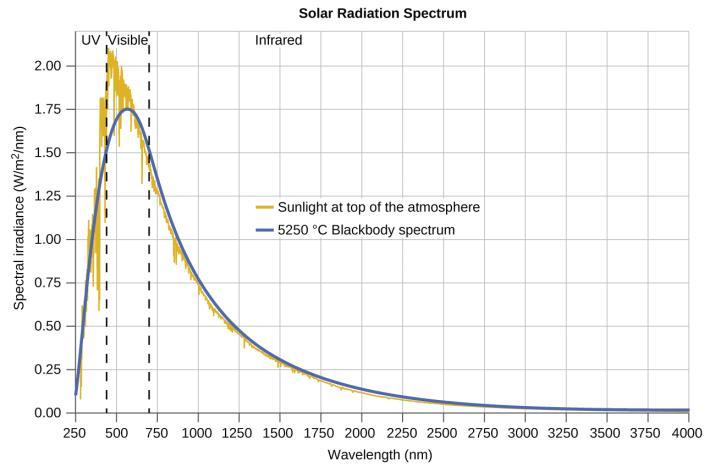
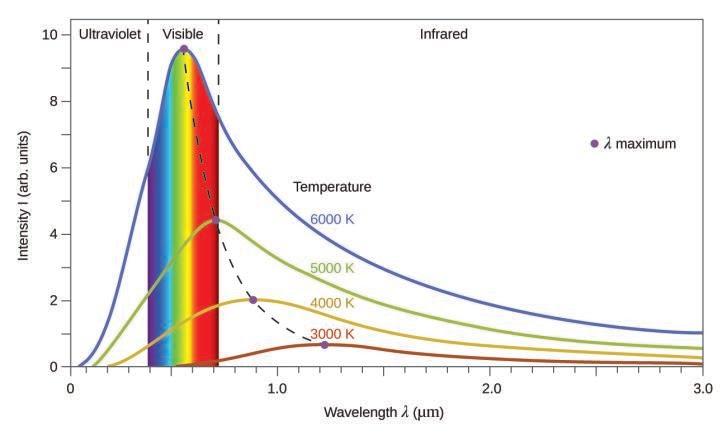
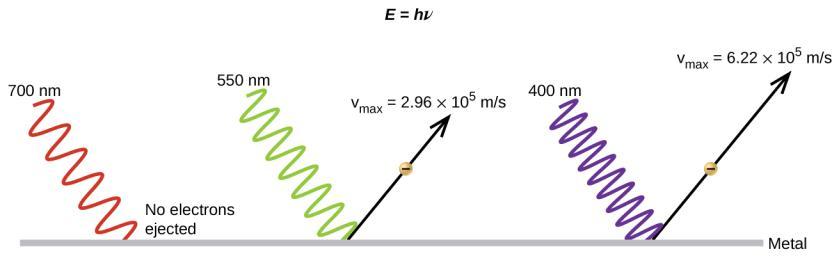

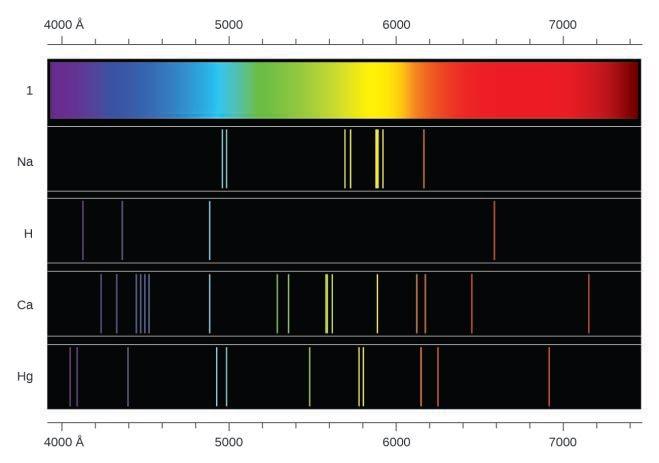
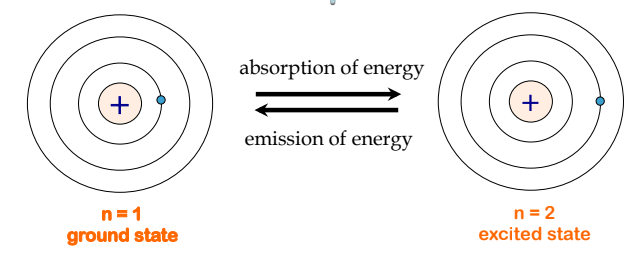
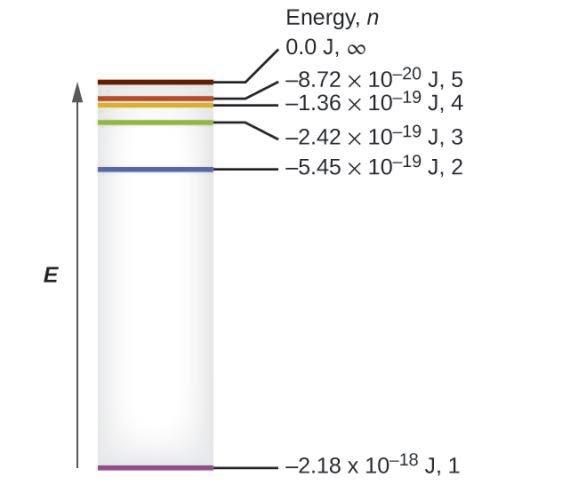
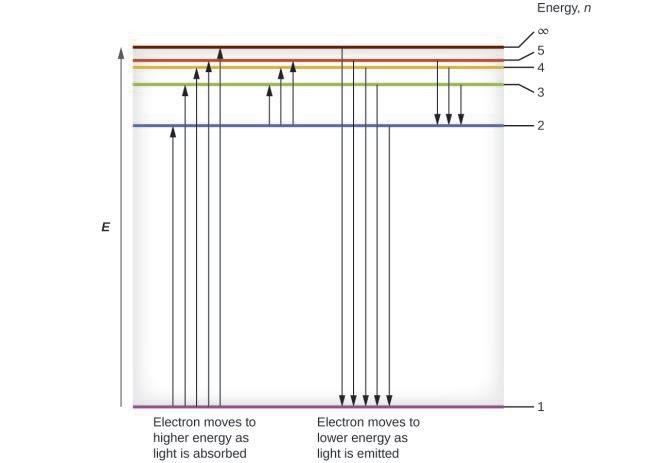
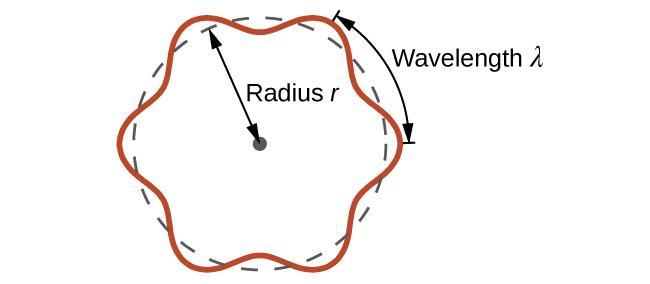
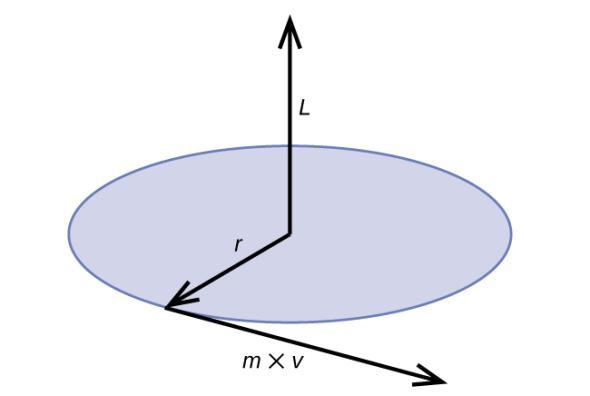
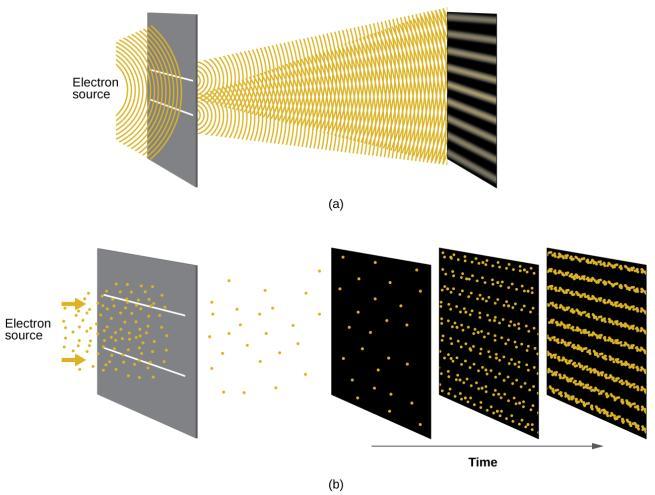
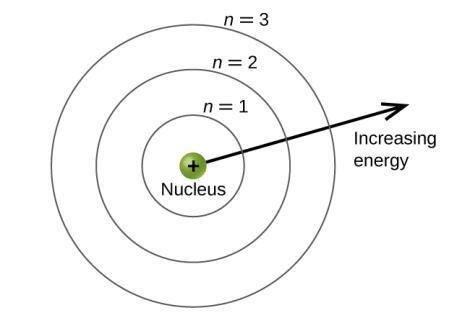
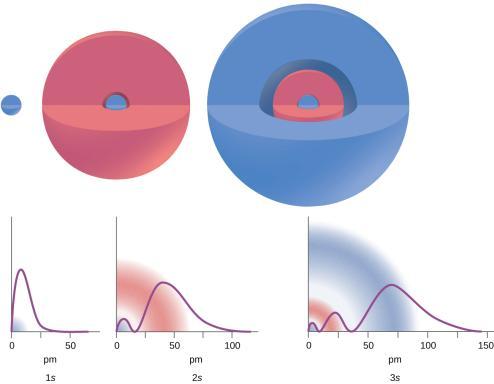
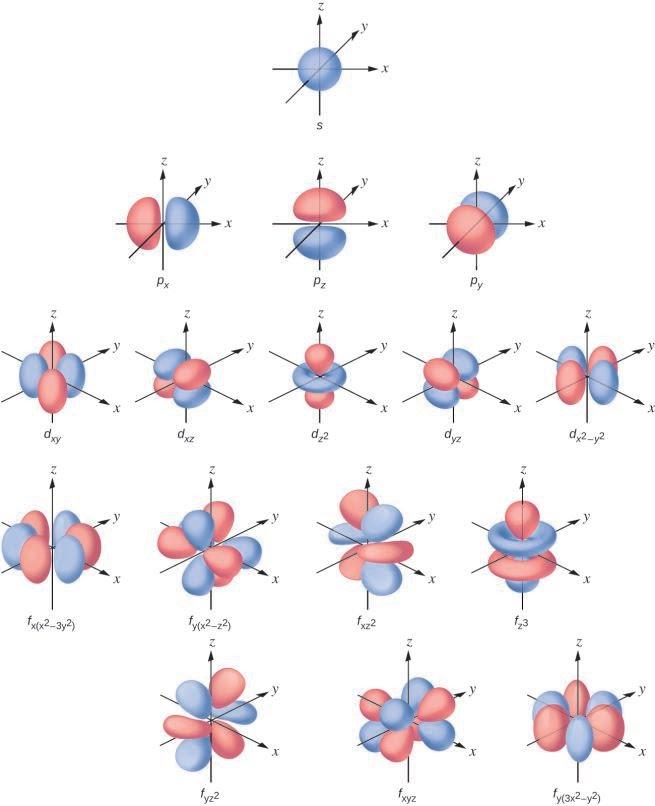
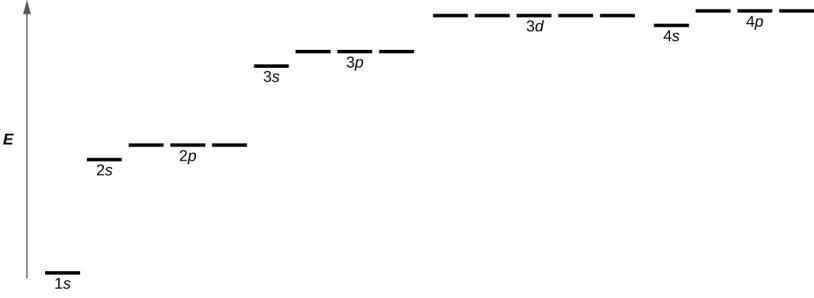
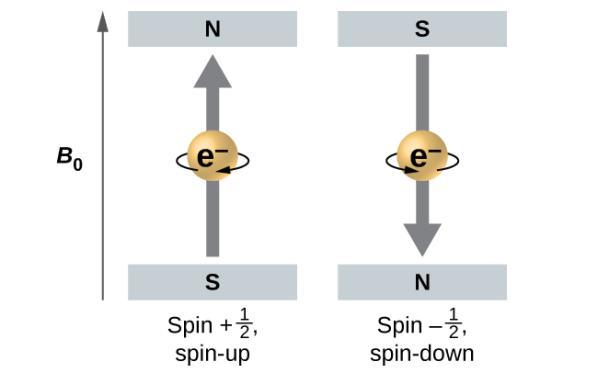
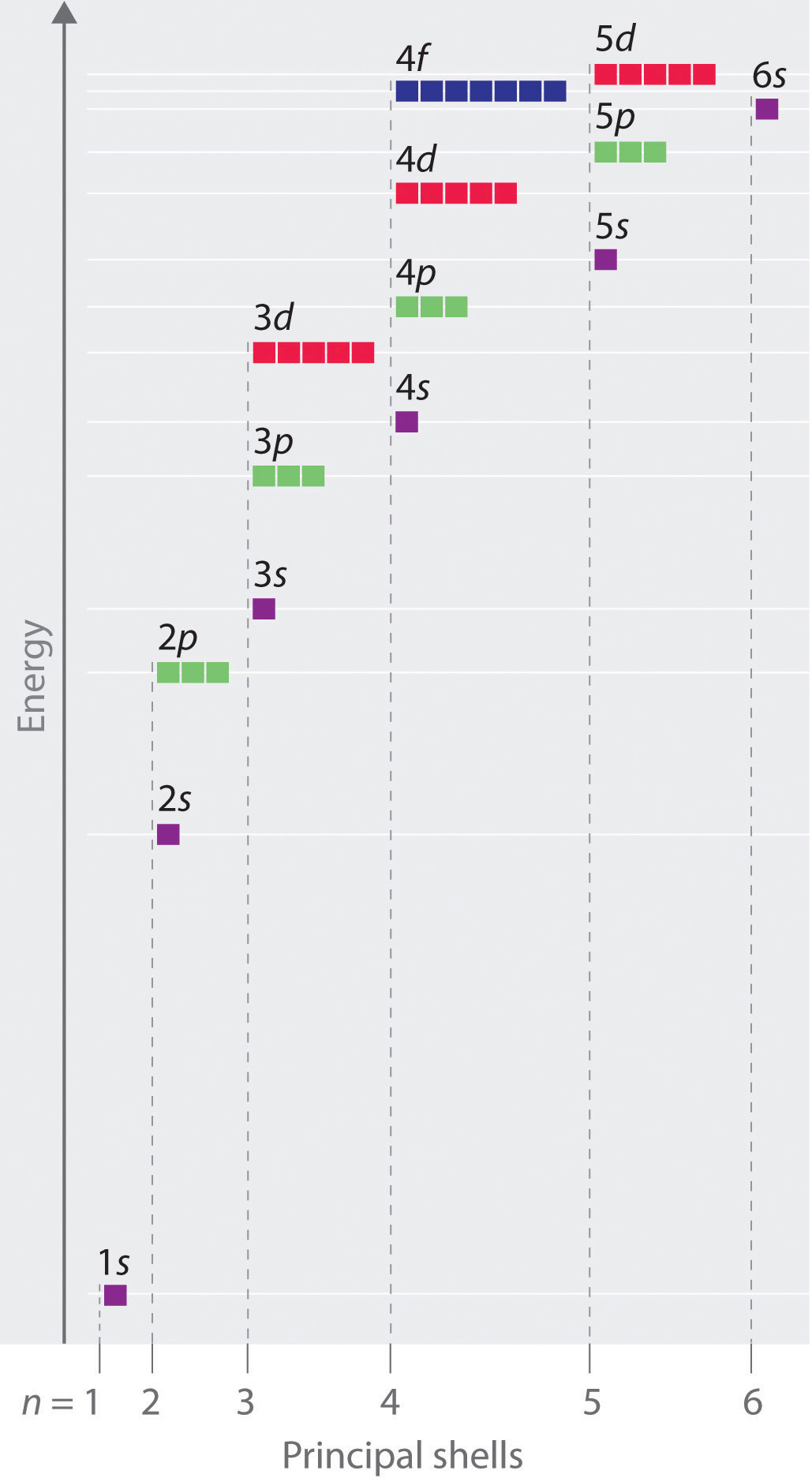
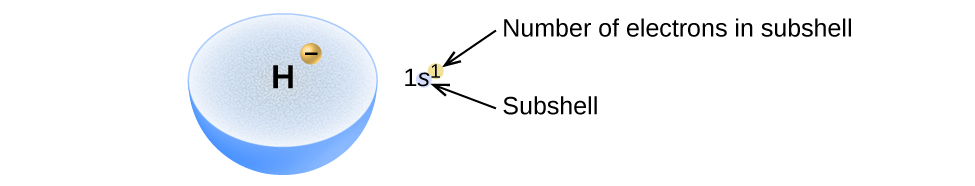
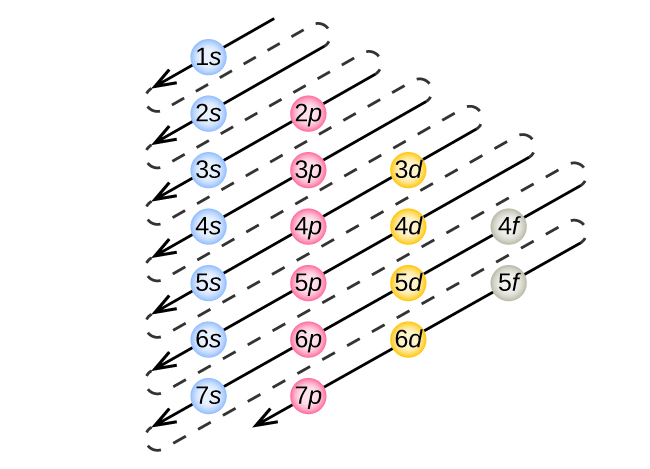
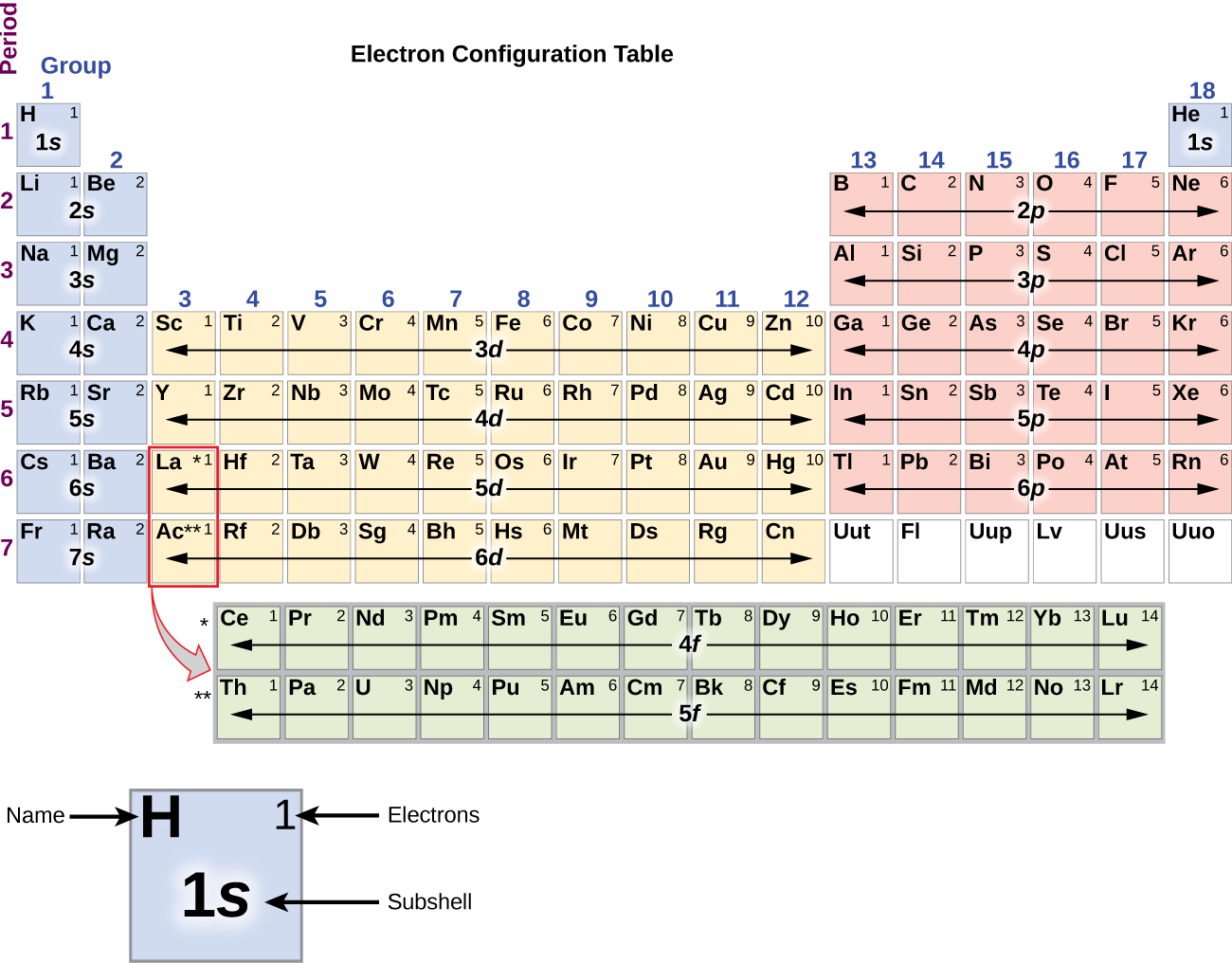


























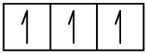
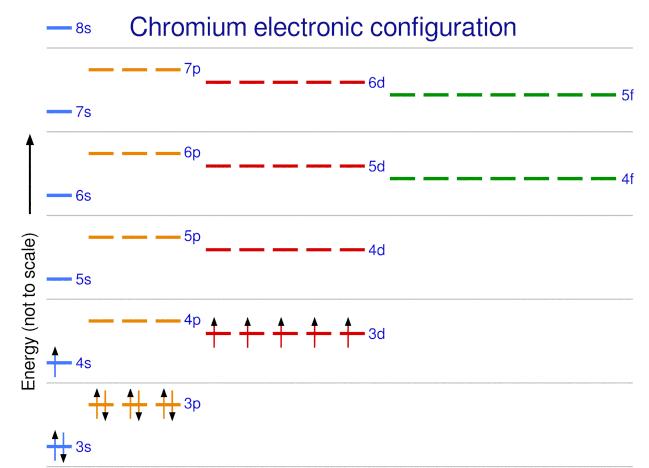
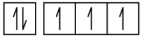
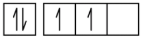


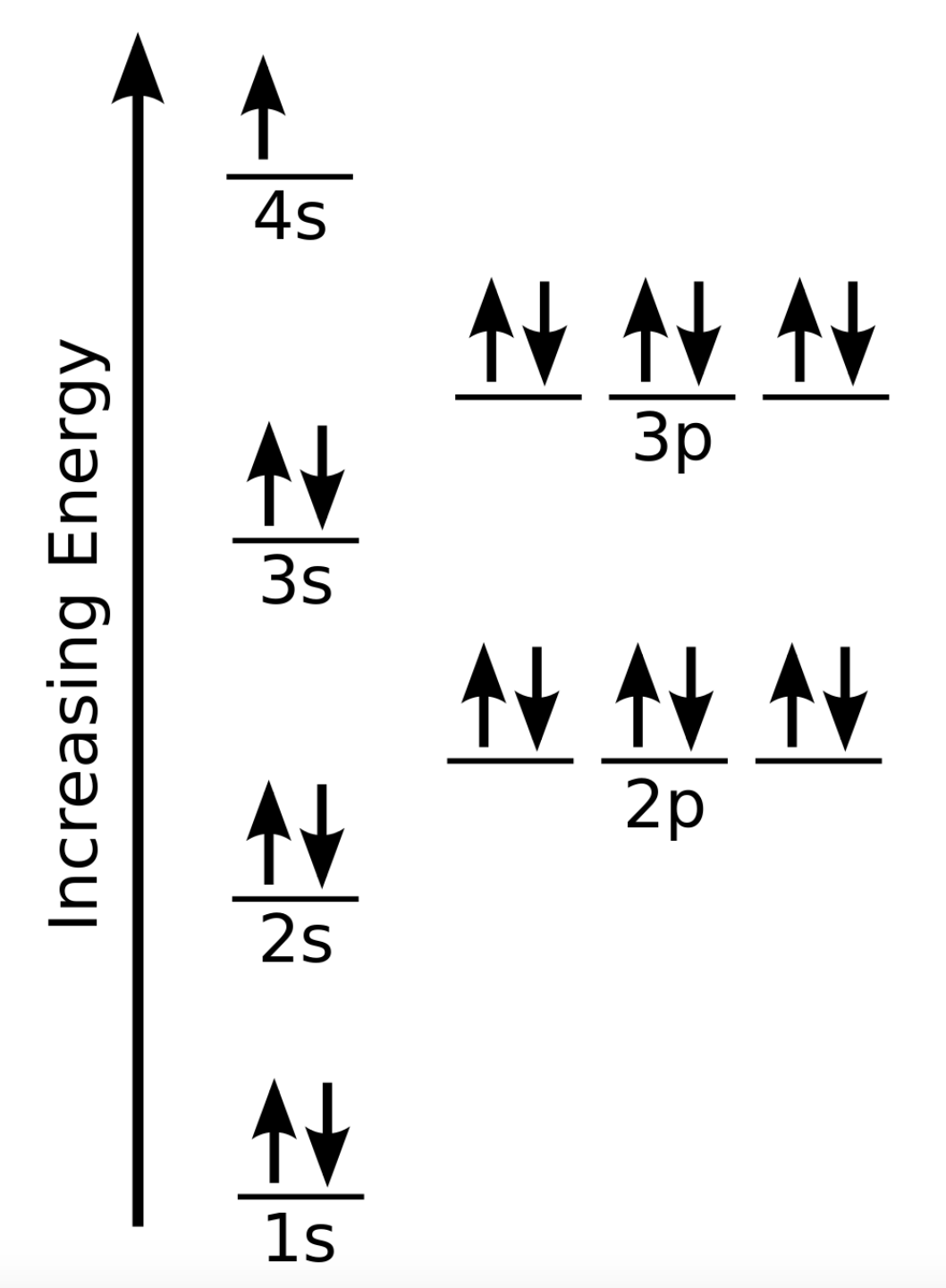

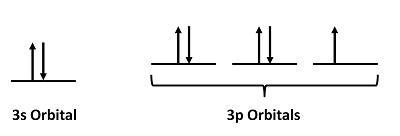
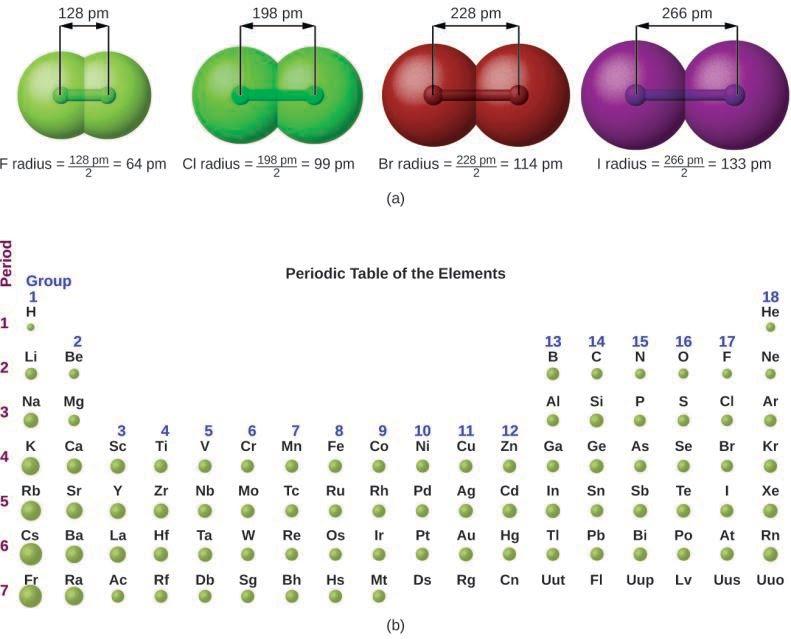
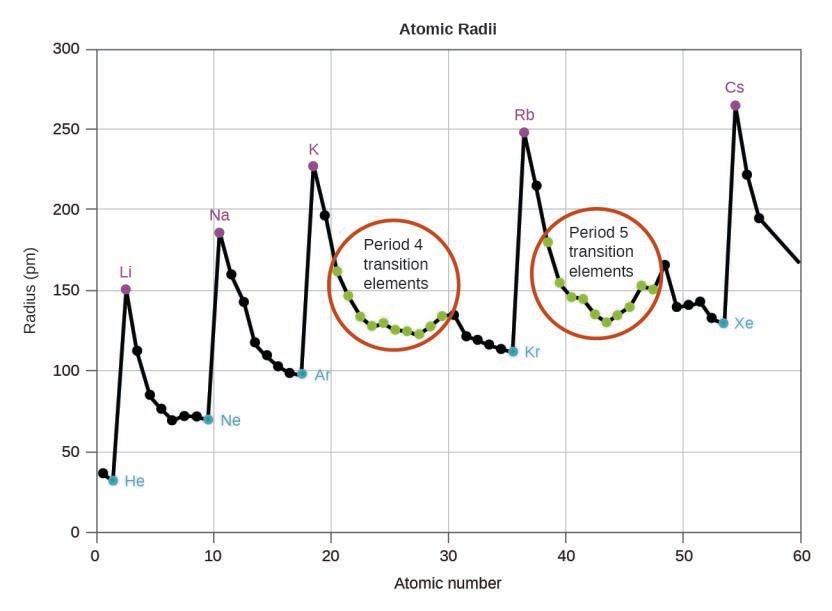

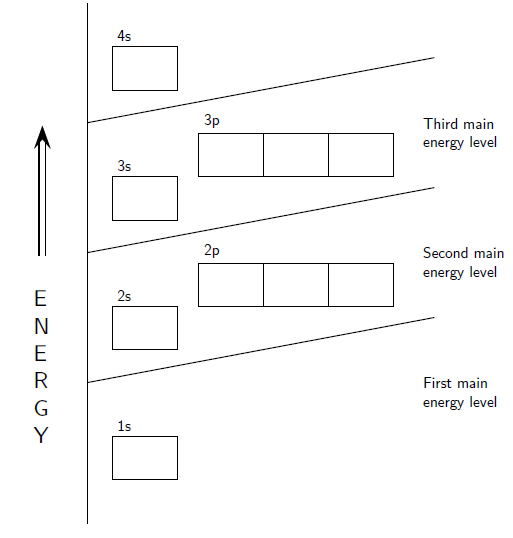
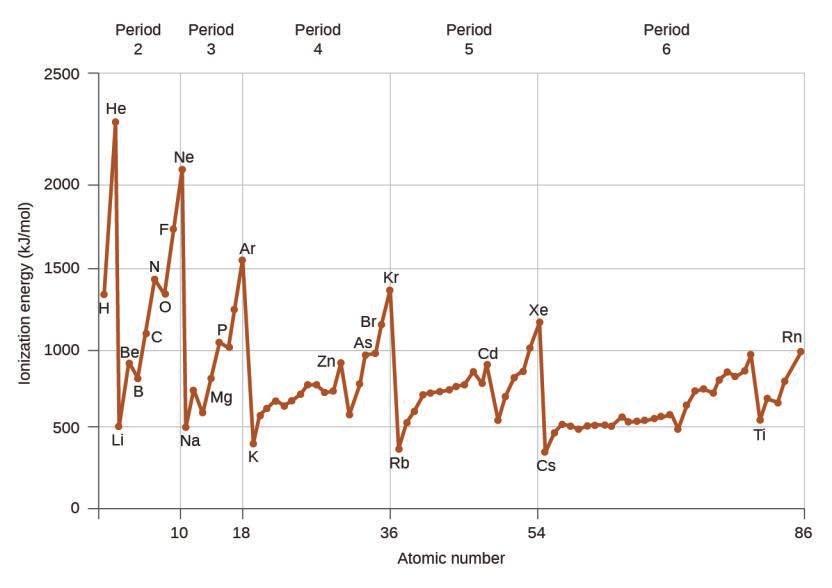
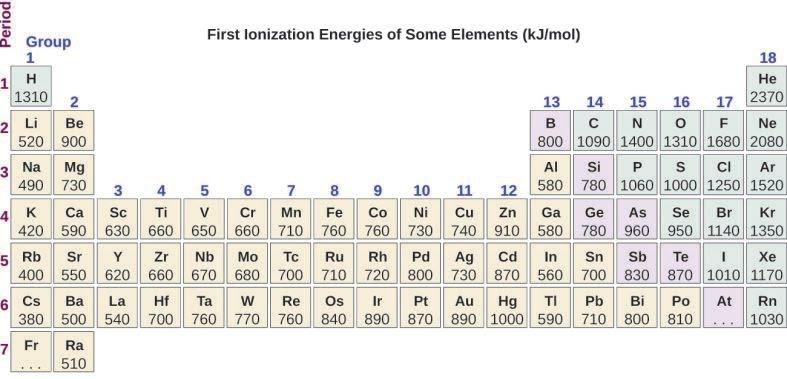
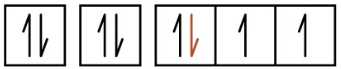
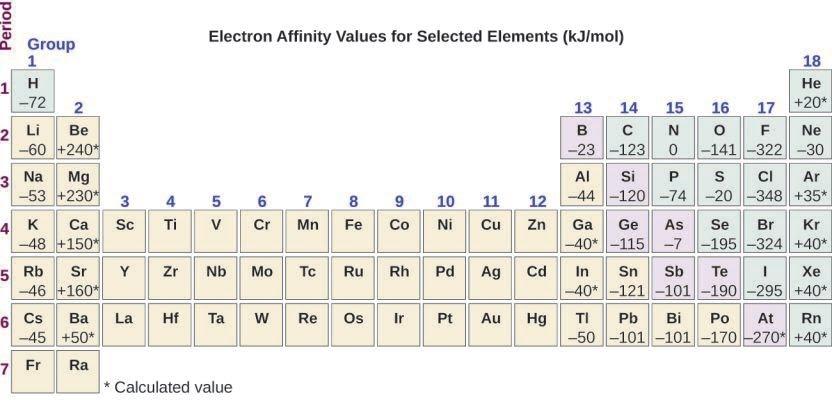
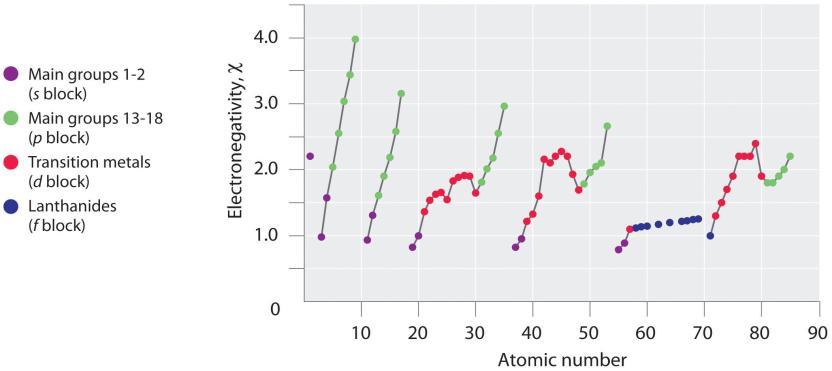
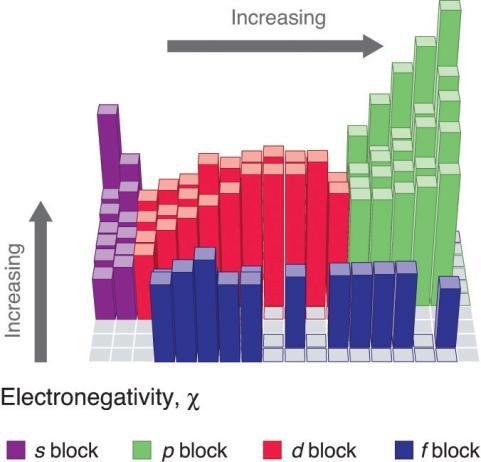
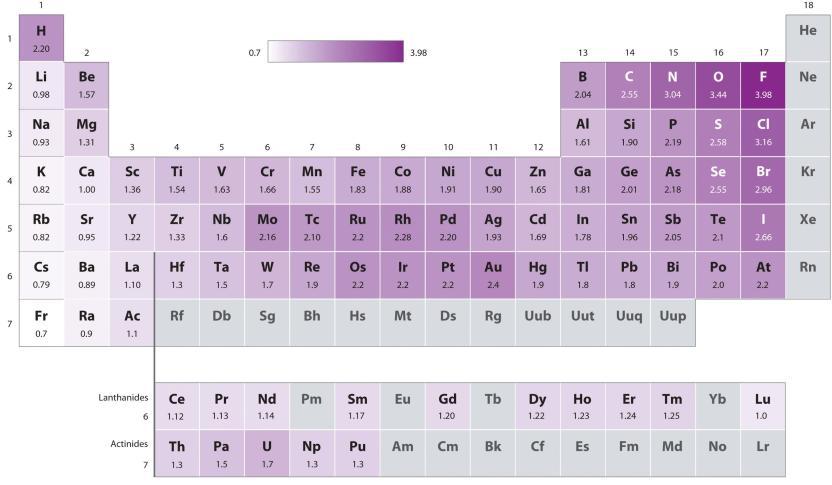
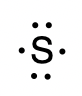
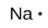
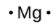
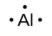
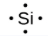
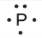
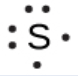
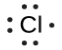


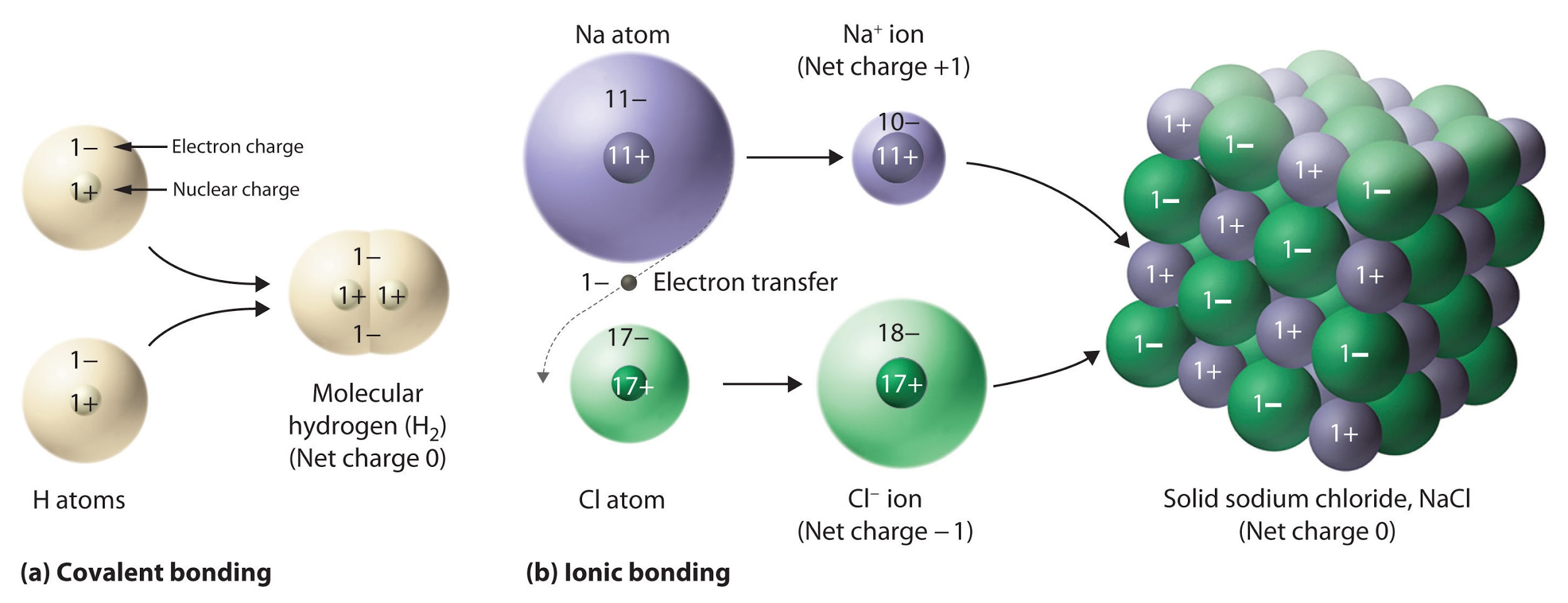
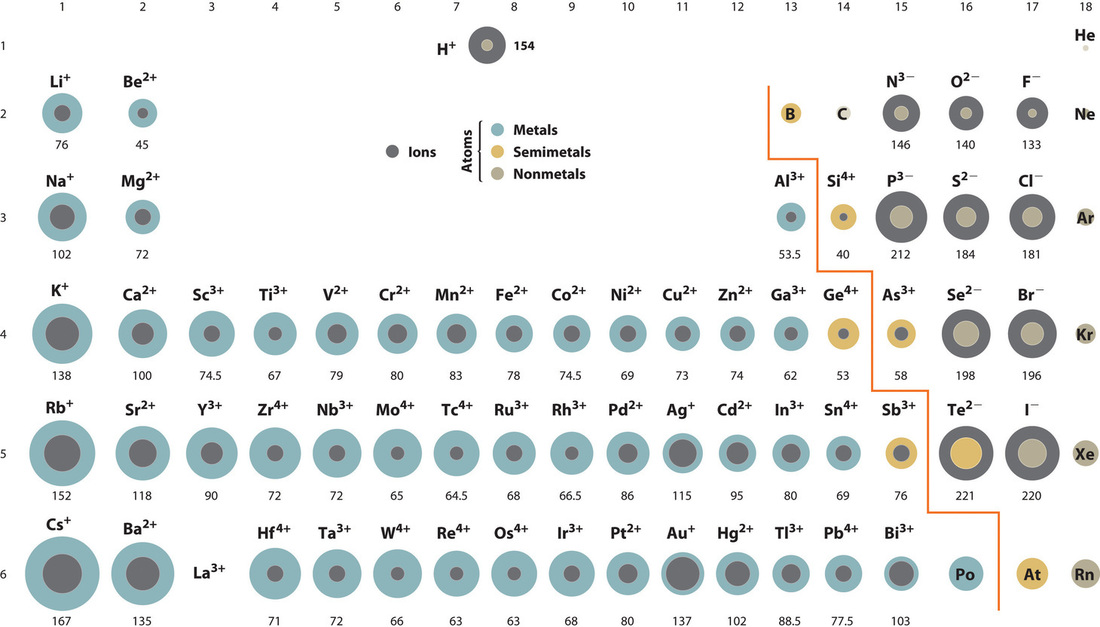
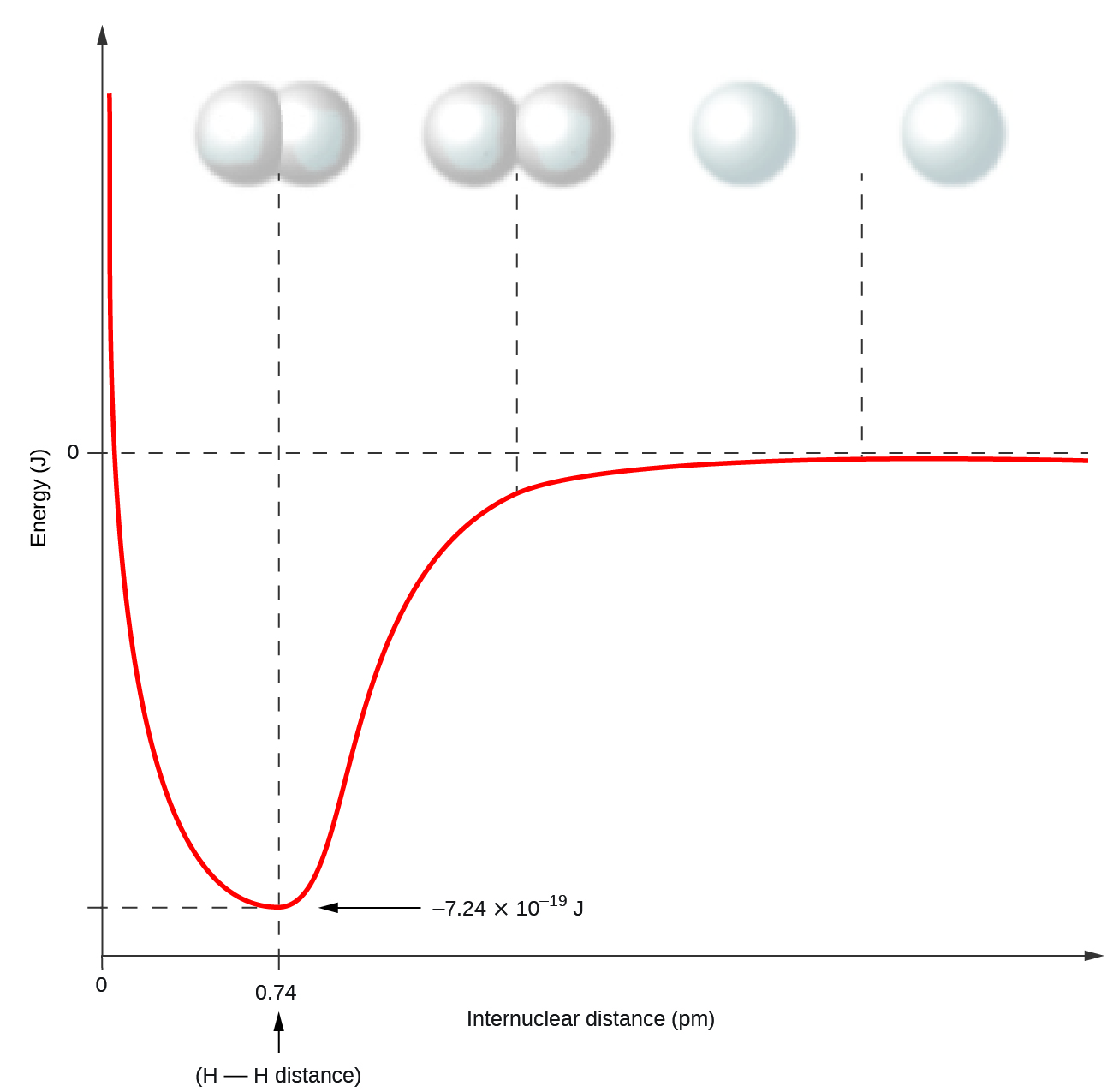
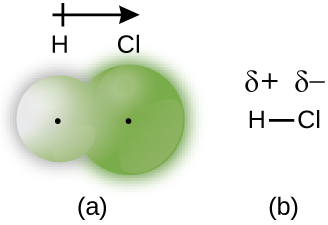

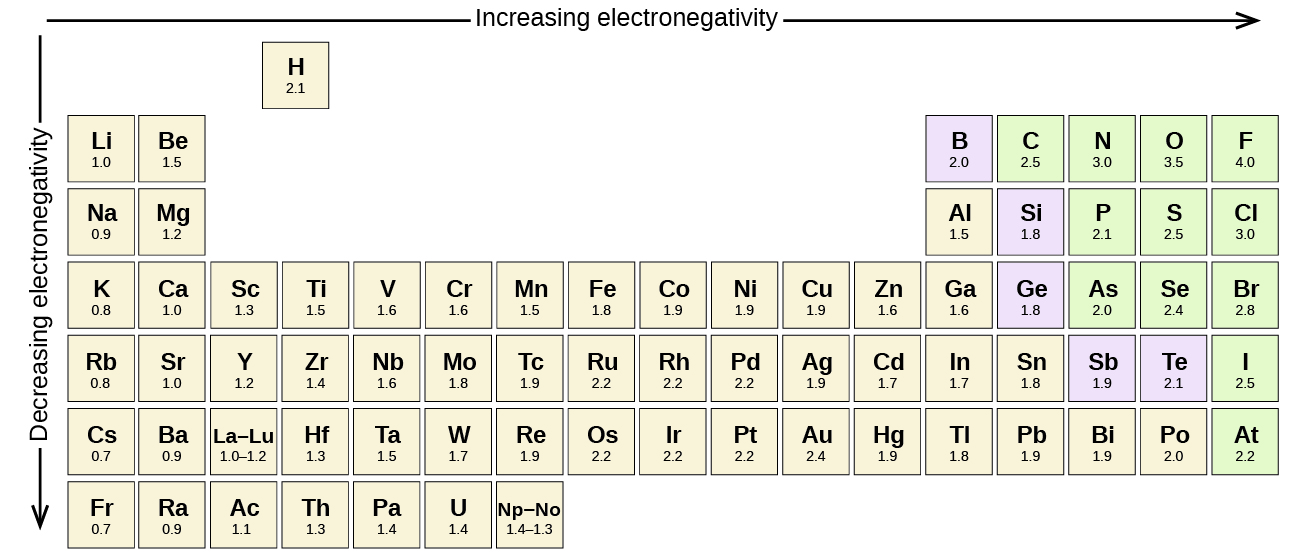
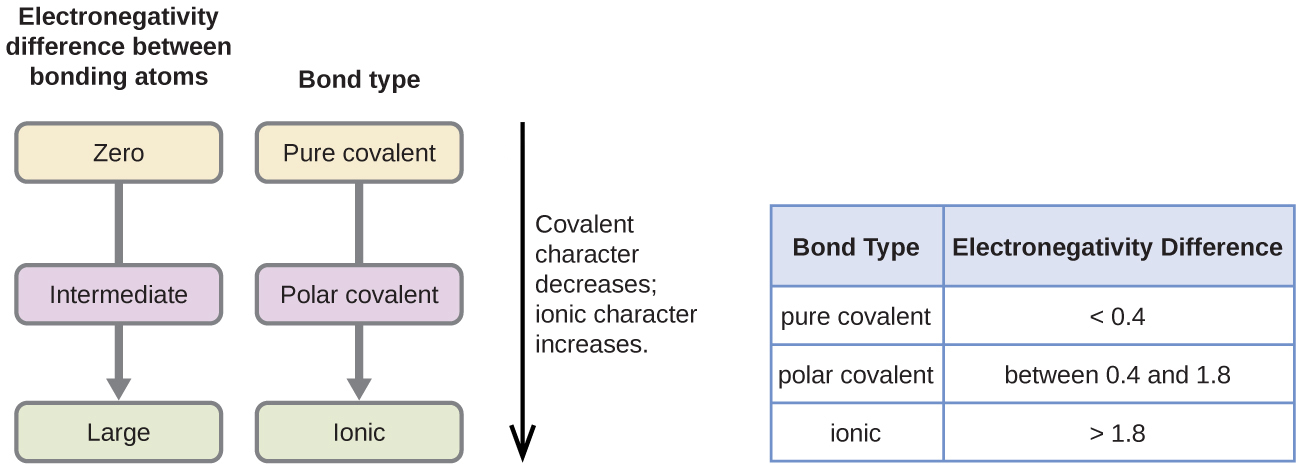
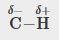
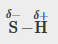
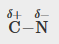
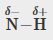
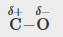
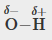
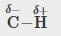
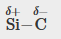
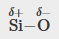

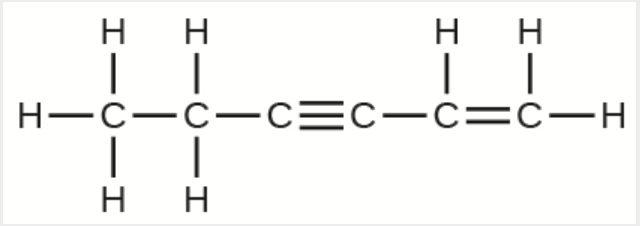


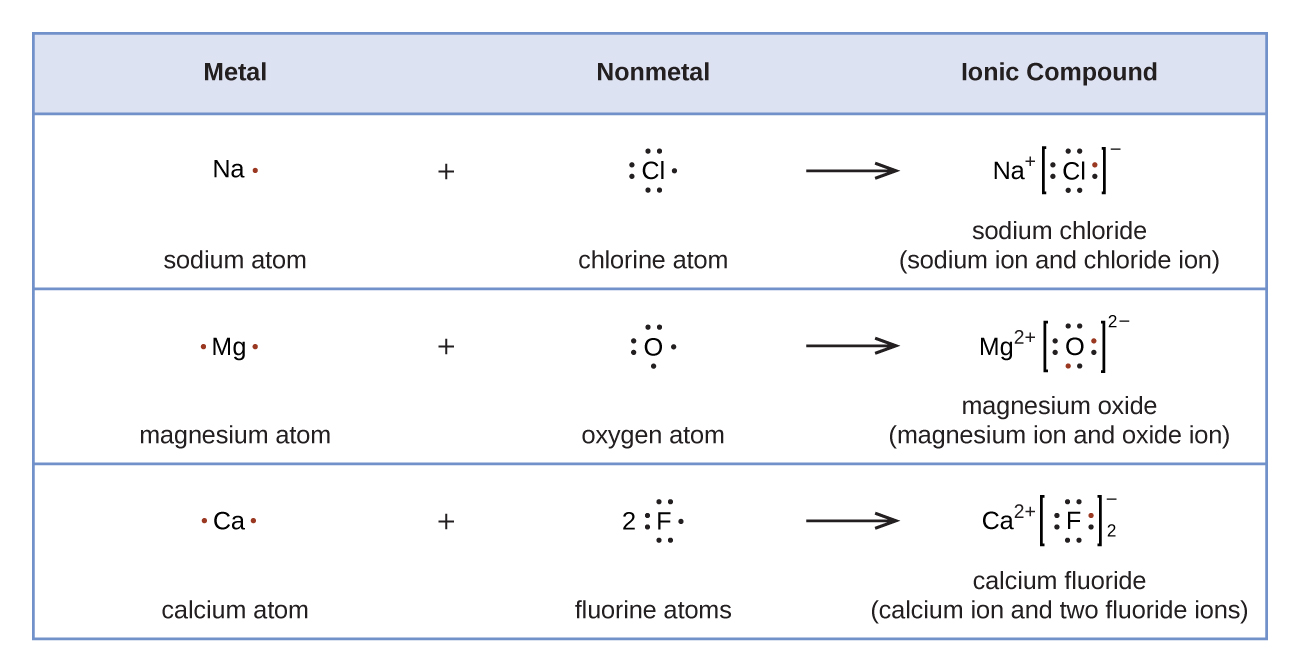
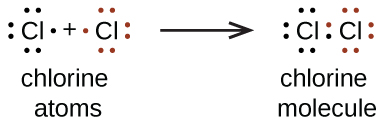

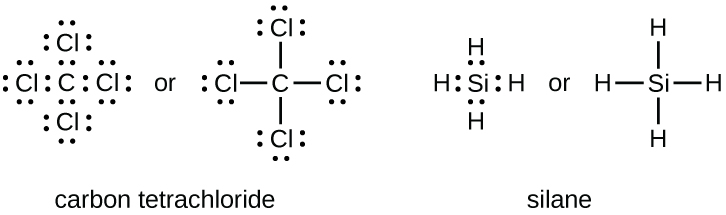
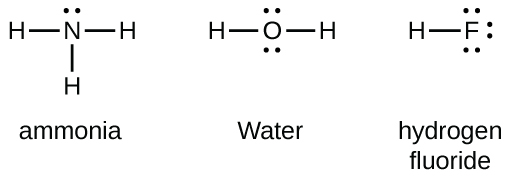
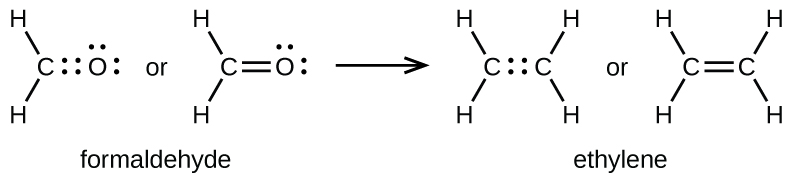
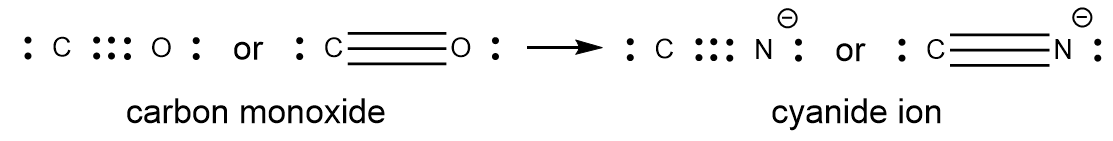
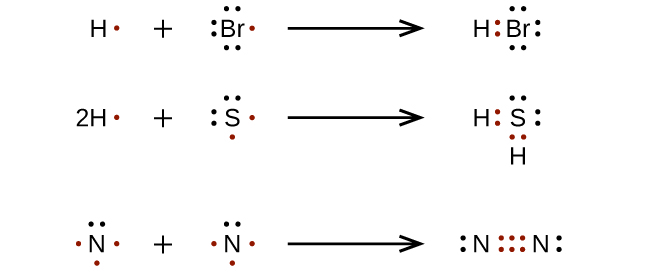
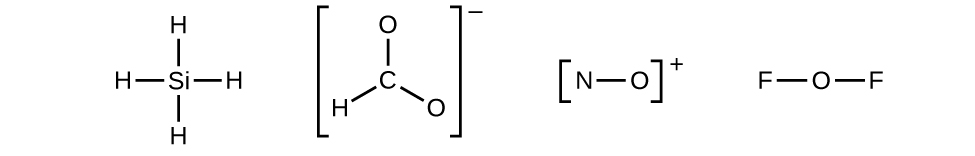
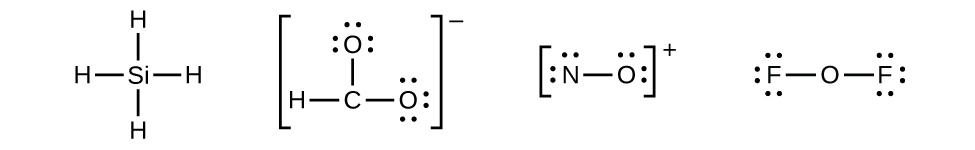
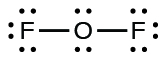
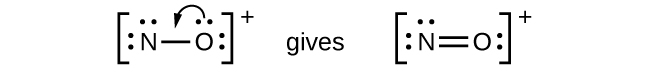
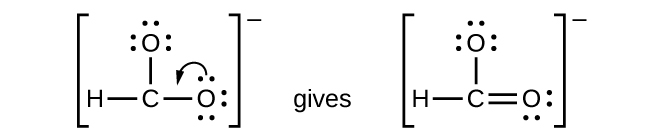
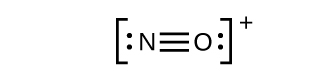
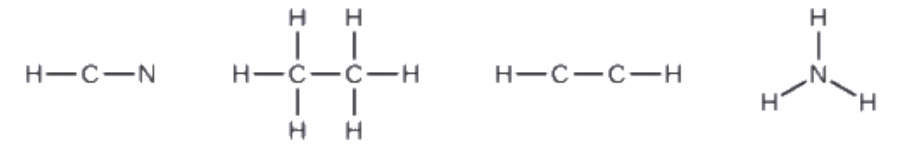
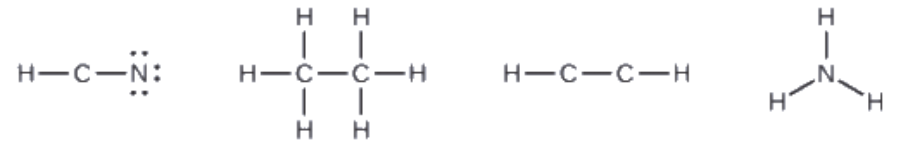
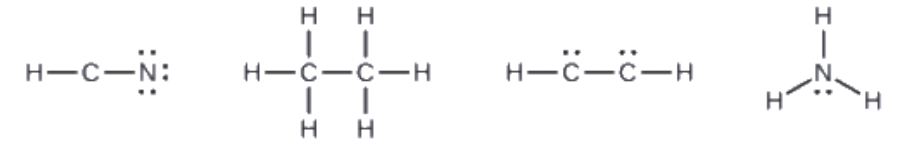
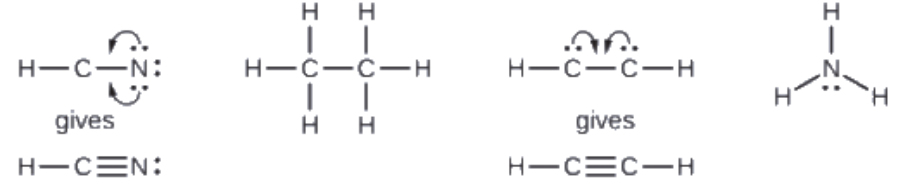
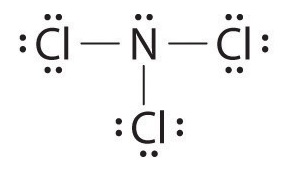
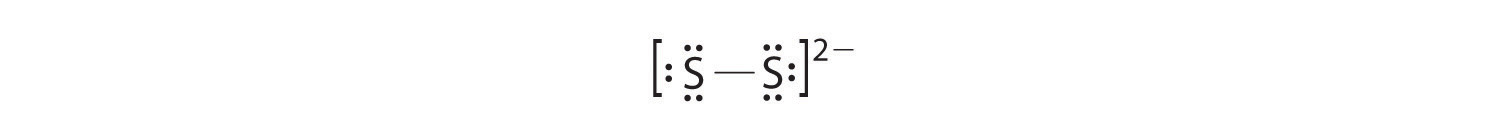
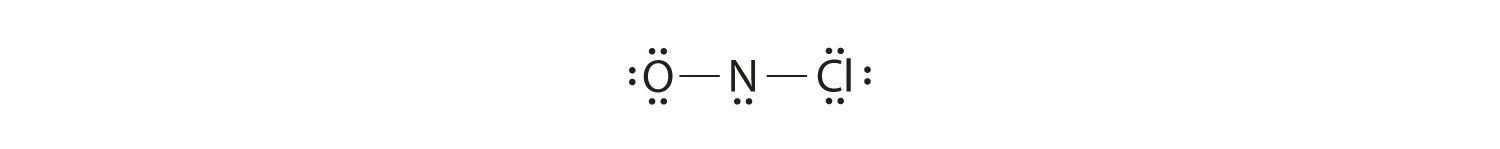
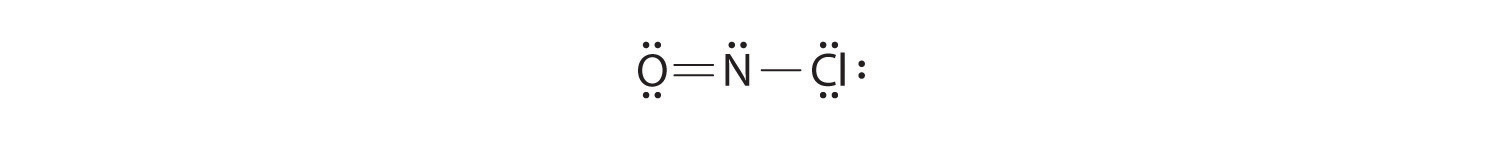
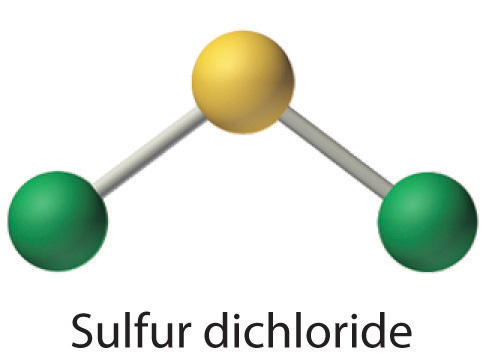
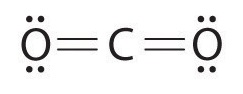
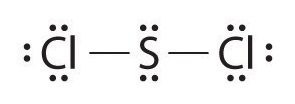

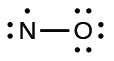
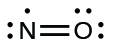
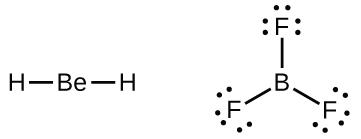
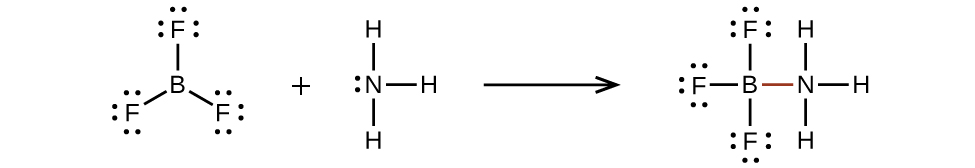
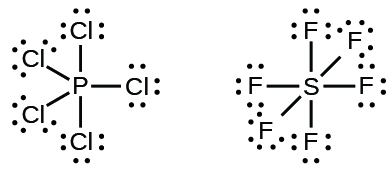
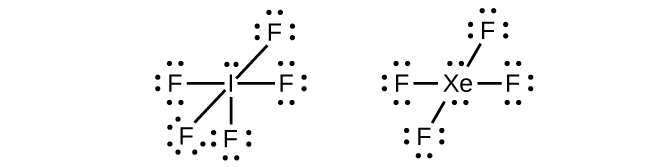
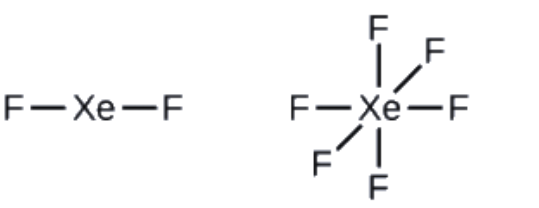
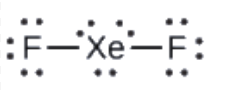
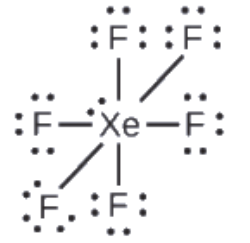
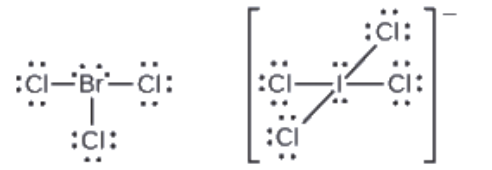
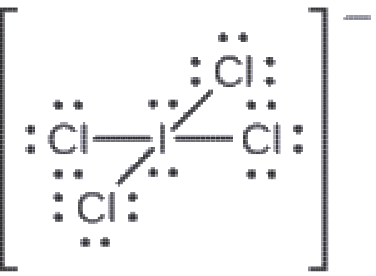
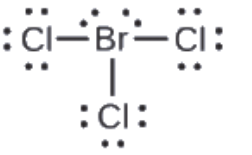
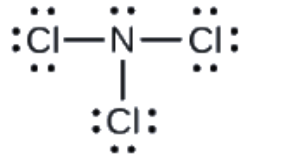
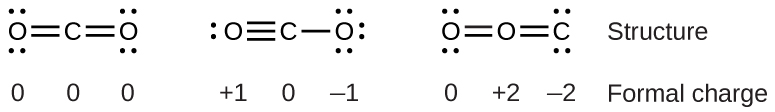
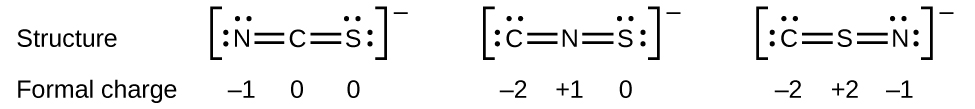



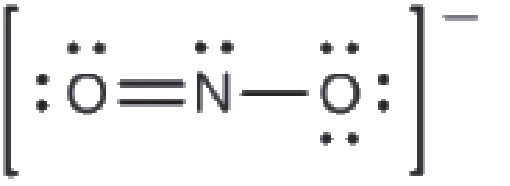
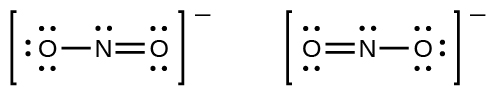
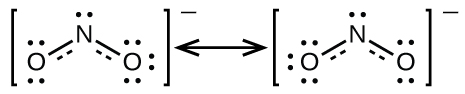
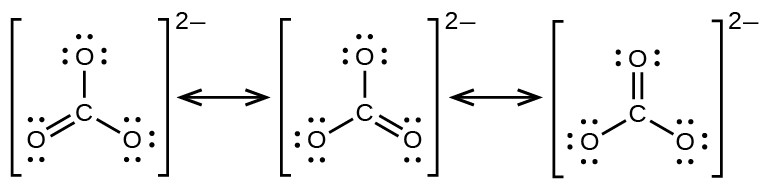
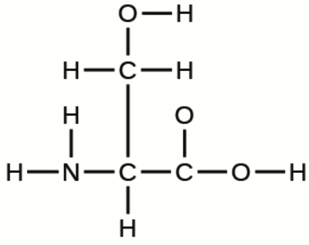
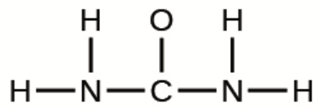
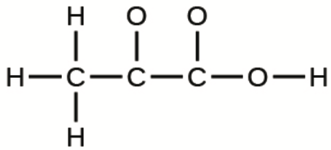
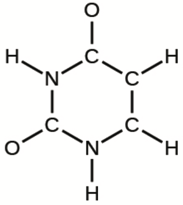
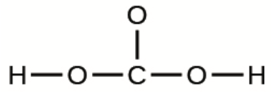
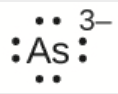

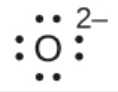
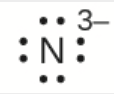






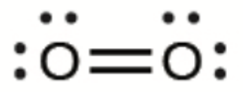
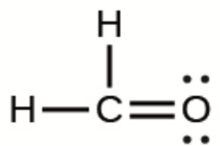
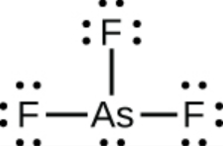
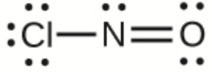
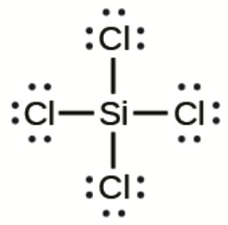
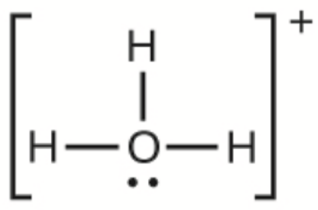
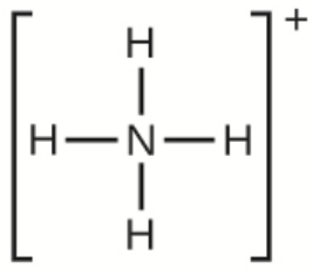
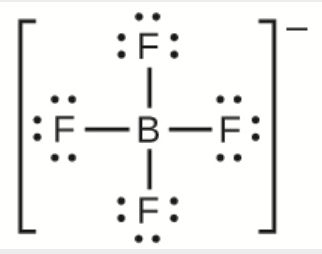

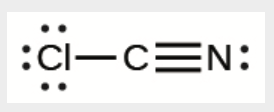
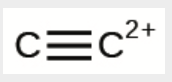
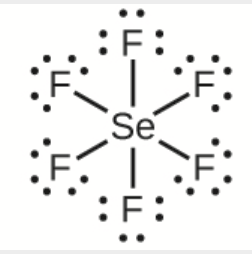
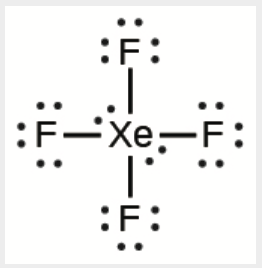
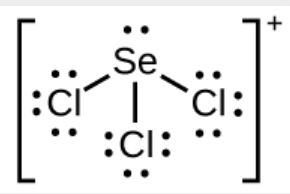
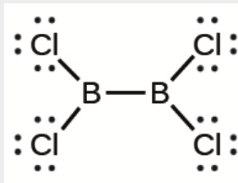

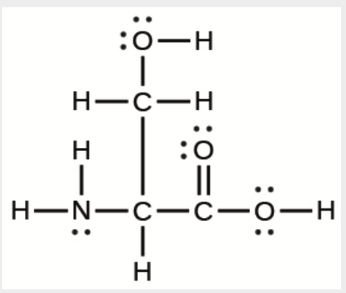
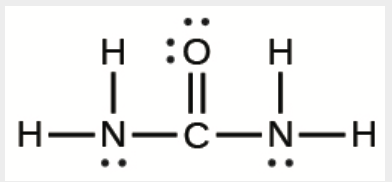
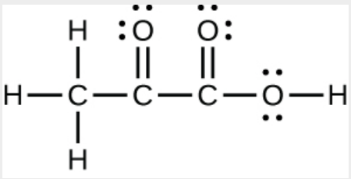
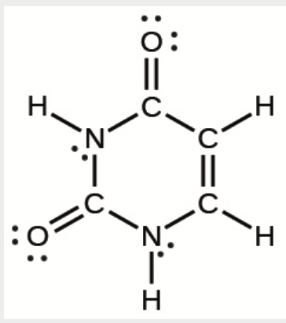
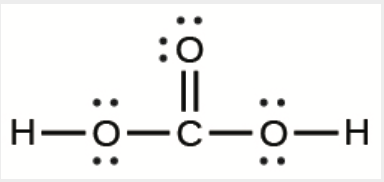
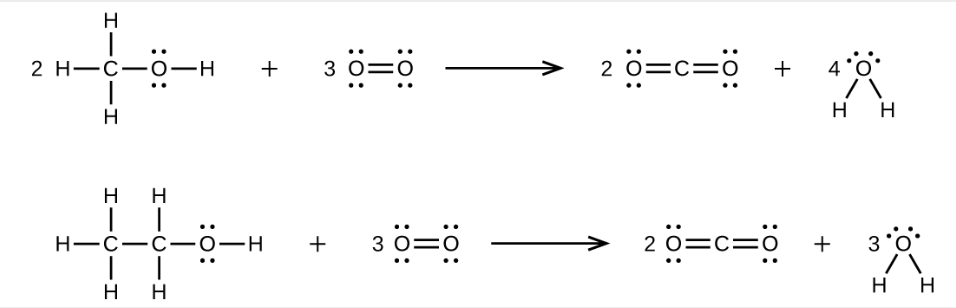
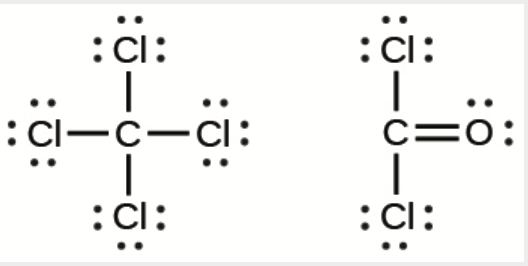
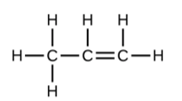
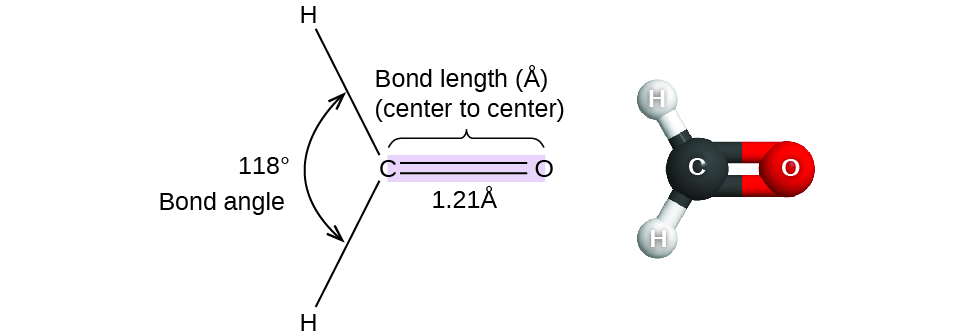
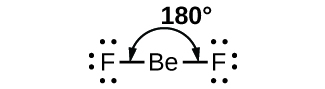
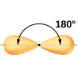
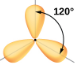
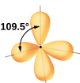
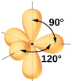
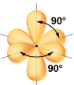
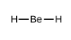
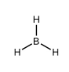
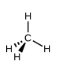
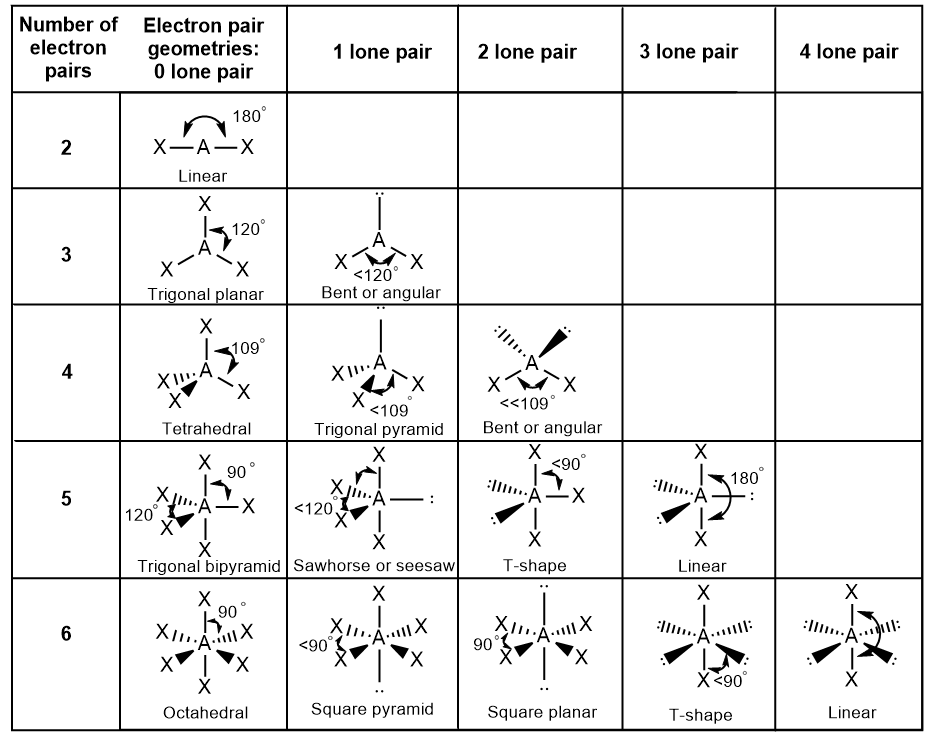
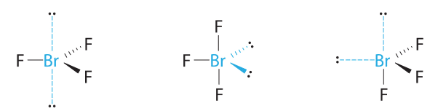
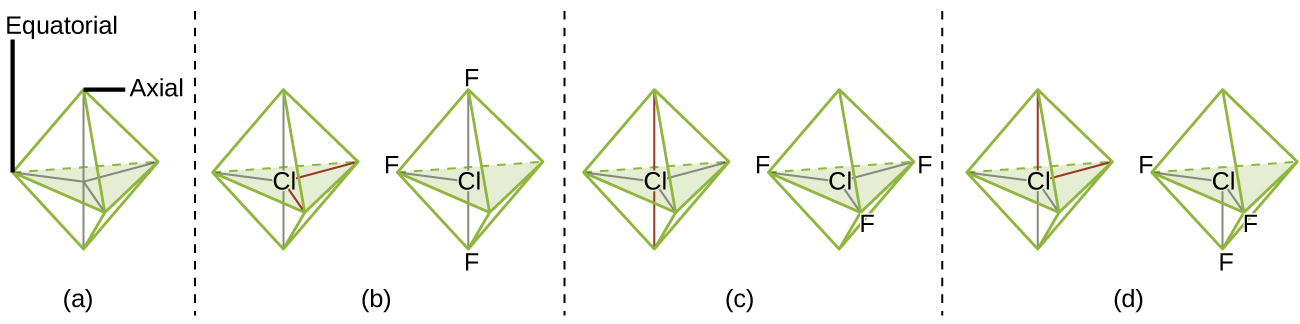
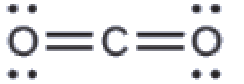
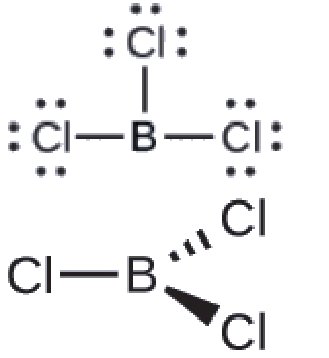
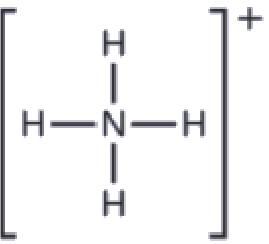
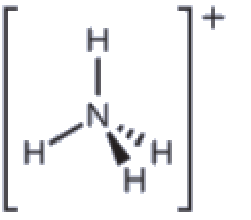
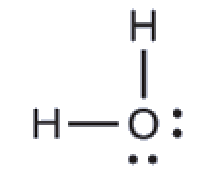

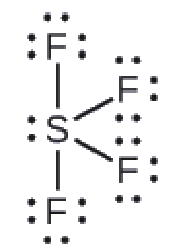
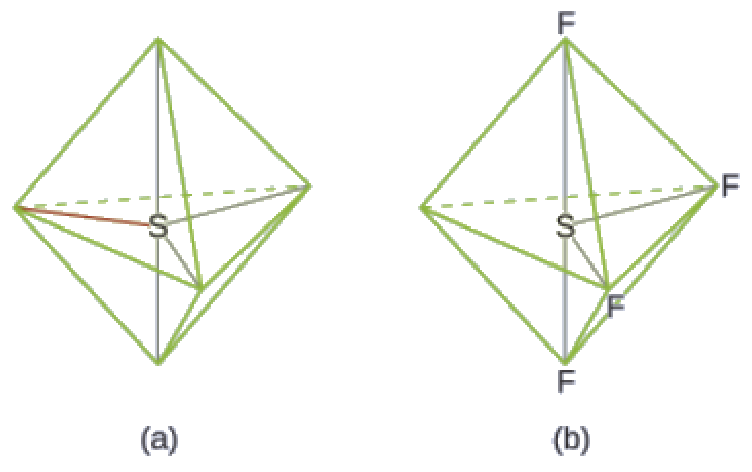
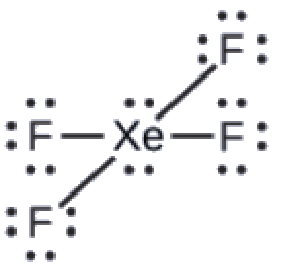
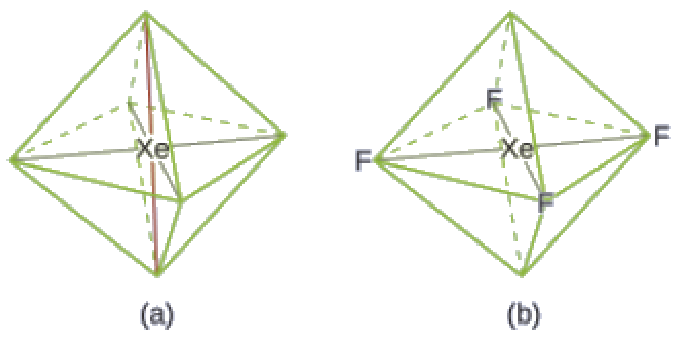
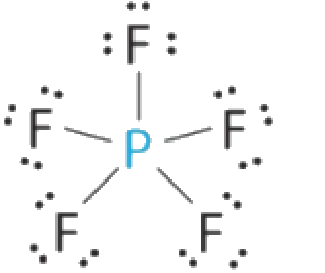
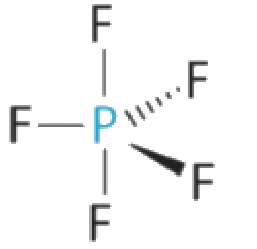
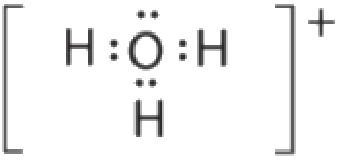
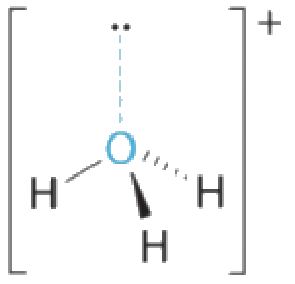
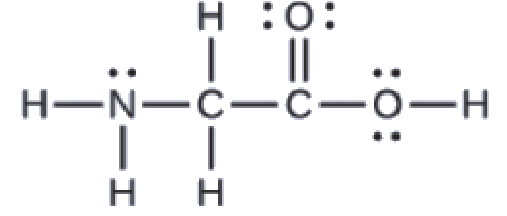
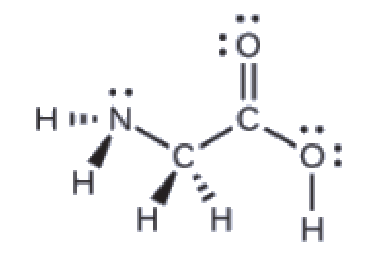
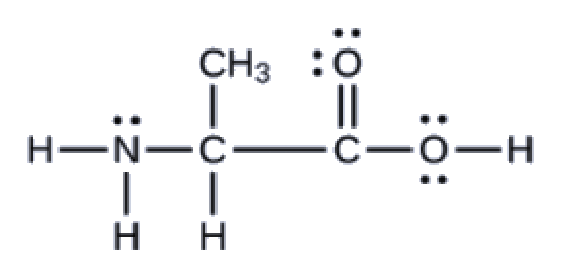
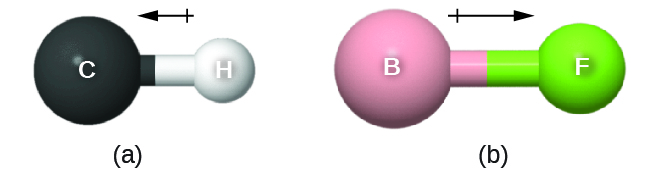
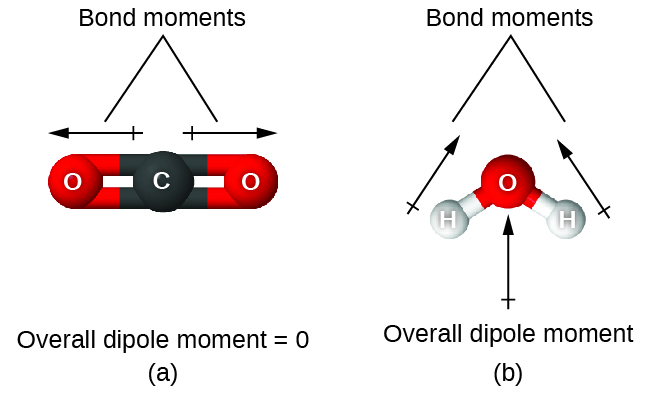
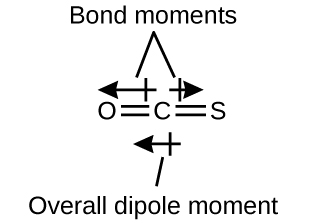
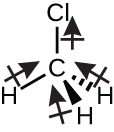
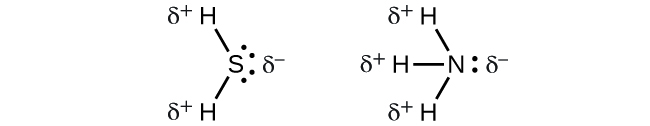
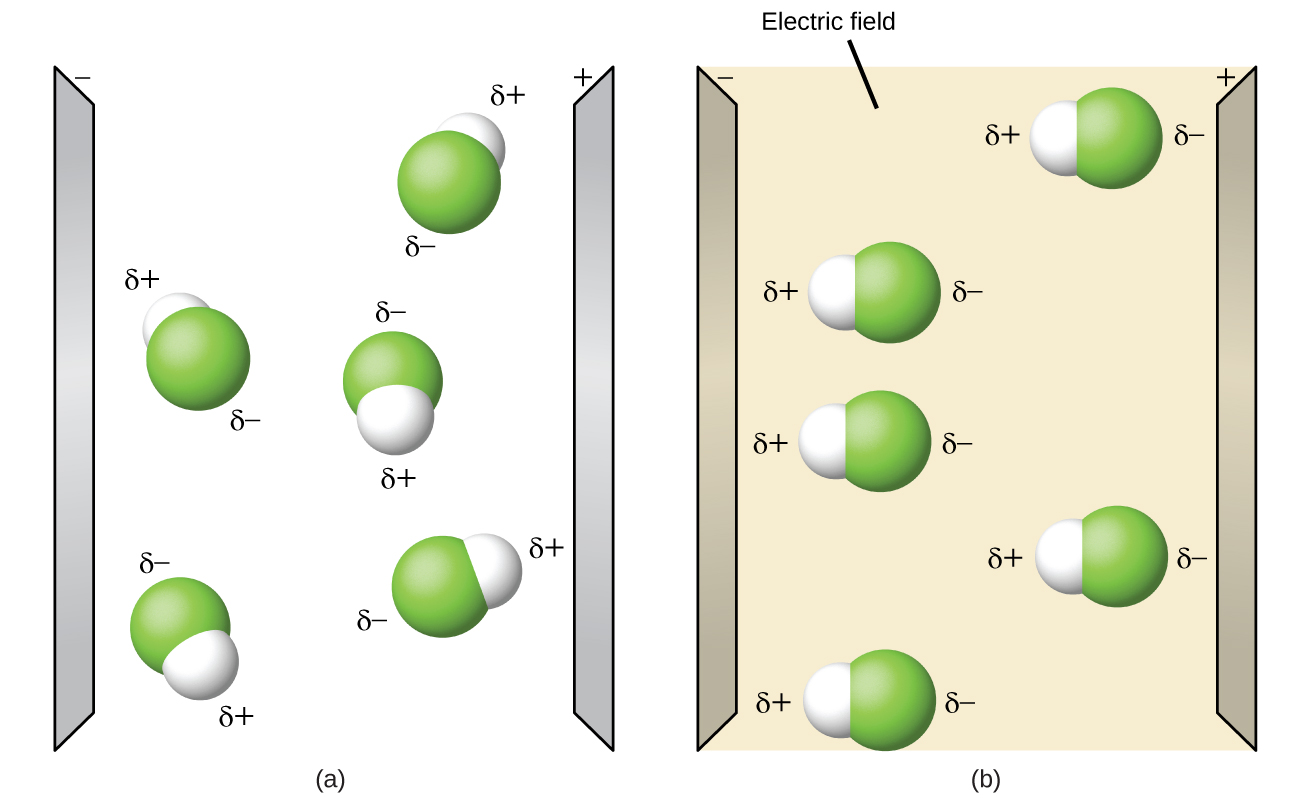
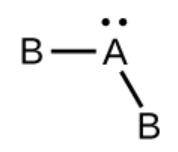
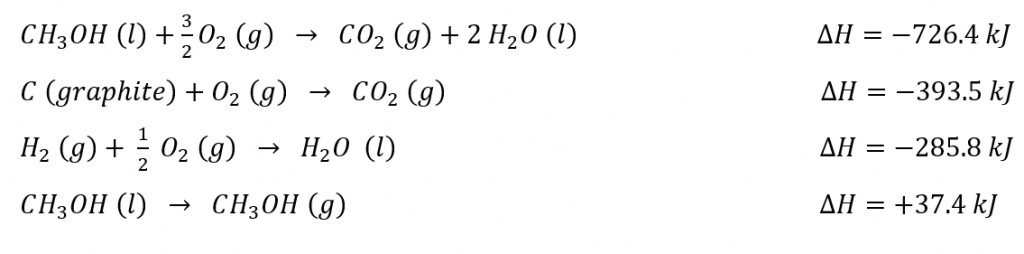
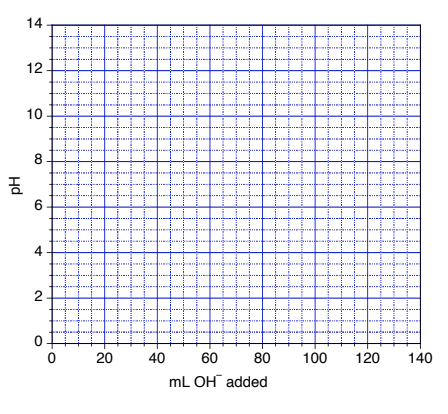
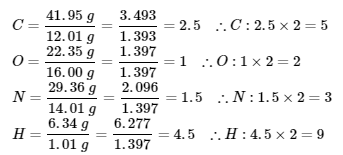

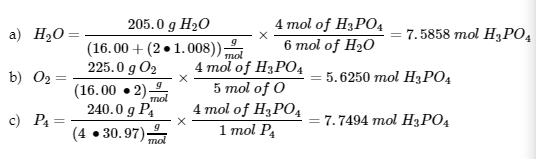
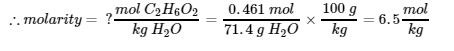

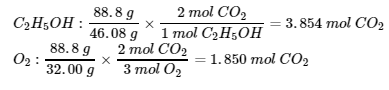


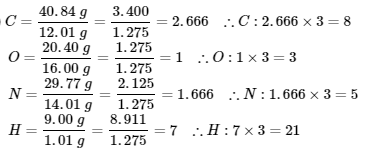
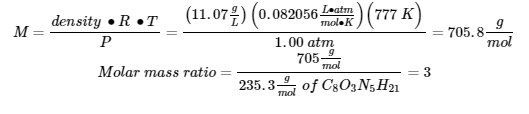
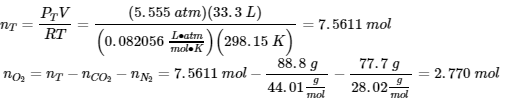

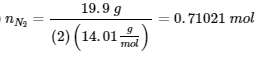

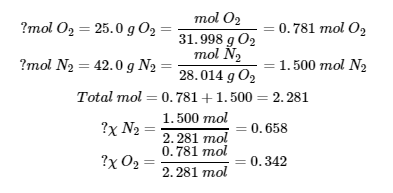
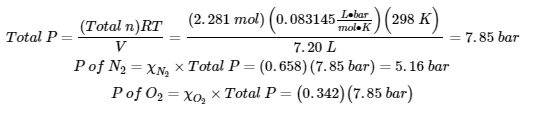
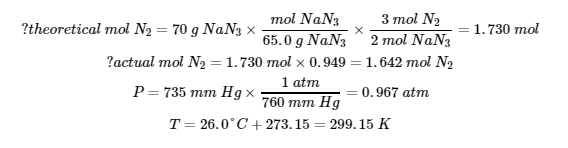
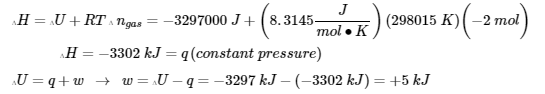

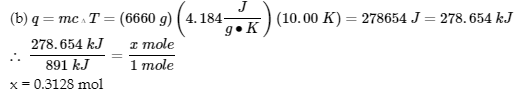


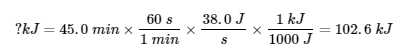
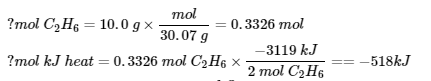
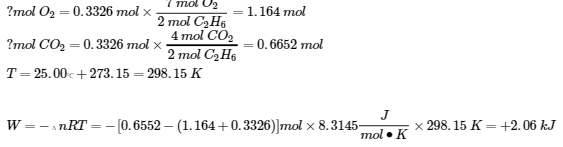
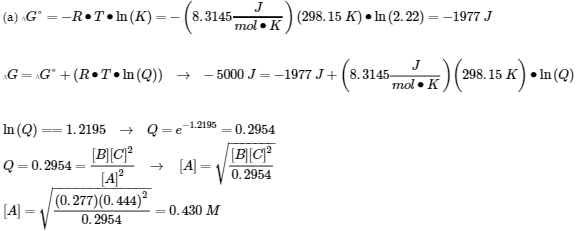
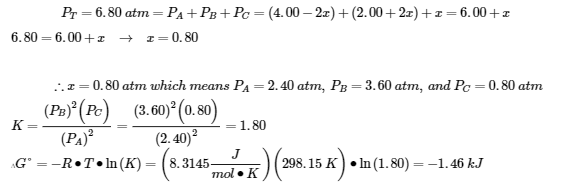
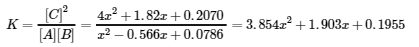
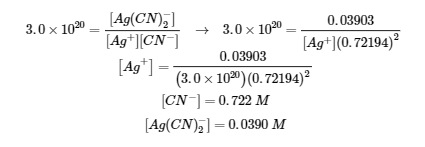
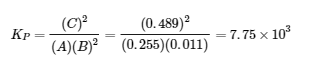
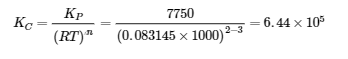
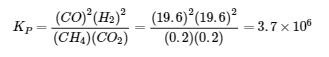
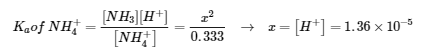
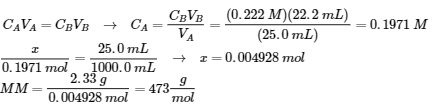
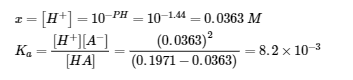
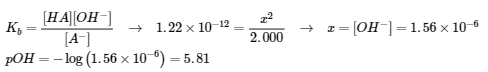
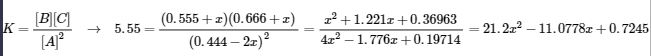
 ∴ pOH = 2.29 and pH = 11.71
∴ pOH = 2.29 and pH = 11.71